6 Traits, 5 tissues, eQTL + sQTL + apaQTL – compute ukbb LD, weights are from Munro et al
XSun
2024-05-27
Last updated: 2024-06-13
Checks: 6 1
Knit directory: multigroup_ctwas_analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20231112) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version c59fd41. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: results/
Unstaged changes:
Modified: analysis/multi_group_6traits_15weights_ukbb_munro.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown
(analysis/multi_group_6traits_15weights_ukbb_munro.Rmd) and
HTML (docs/multi_group_6traits_15weights_ukbb_munro.html)
files. If you’ve configured a remote Git repository (see
?wflow_git_remote), click on the hyperlinks in the table
below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 92a8e35 | XSun | 2024-06-13 | update |
| html | 92a8e35 | XSun | 2024-06-13 | update |
| Rmd | 9e83189 | XSun | 2024-06-13 | update |
| html | 9e83189 | XSun | 2024-06-13 | update |
Overview
Tissues
The independent tissues are selected by single tissue analysis
Omics
eQTL, sQTL, apaQTL weights are from Munro et al.
Settings
- Weight processing:
PredictDB:
all the PredictDB are converted from FUSION weights
- drop_strand_ambig = TRUE,
- scale_by_ld_variance = F (FUSION converted weights)
- load_predictdb_LD = F,
- Parameter estimation and fine-mapping
- niter_prefit = 5,
- niter = 60,
- L = 3,
- group_prior_var_structure = “shared_type”,
- maxSNP = 20000,
- min_nonSNP_PIP = 0.5,
- Memory requested
- cpus-per-task=2
- mem=200G
over 30h running time
Results
Results from multi-group analysis
The results are summarized by
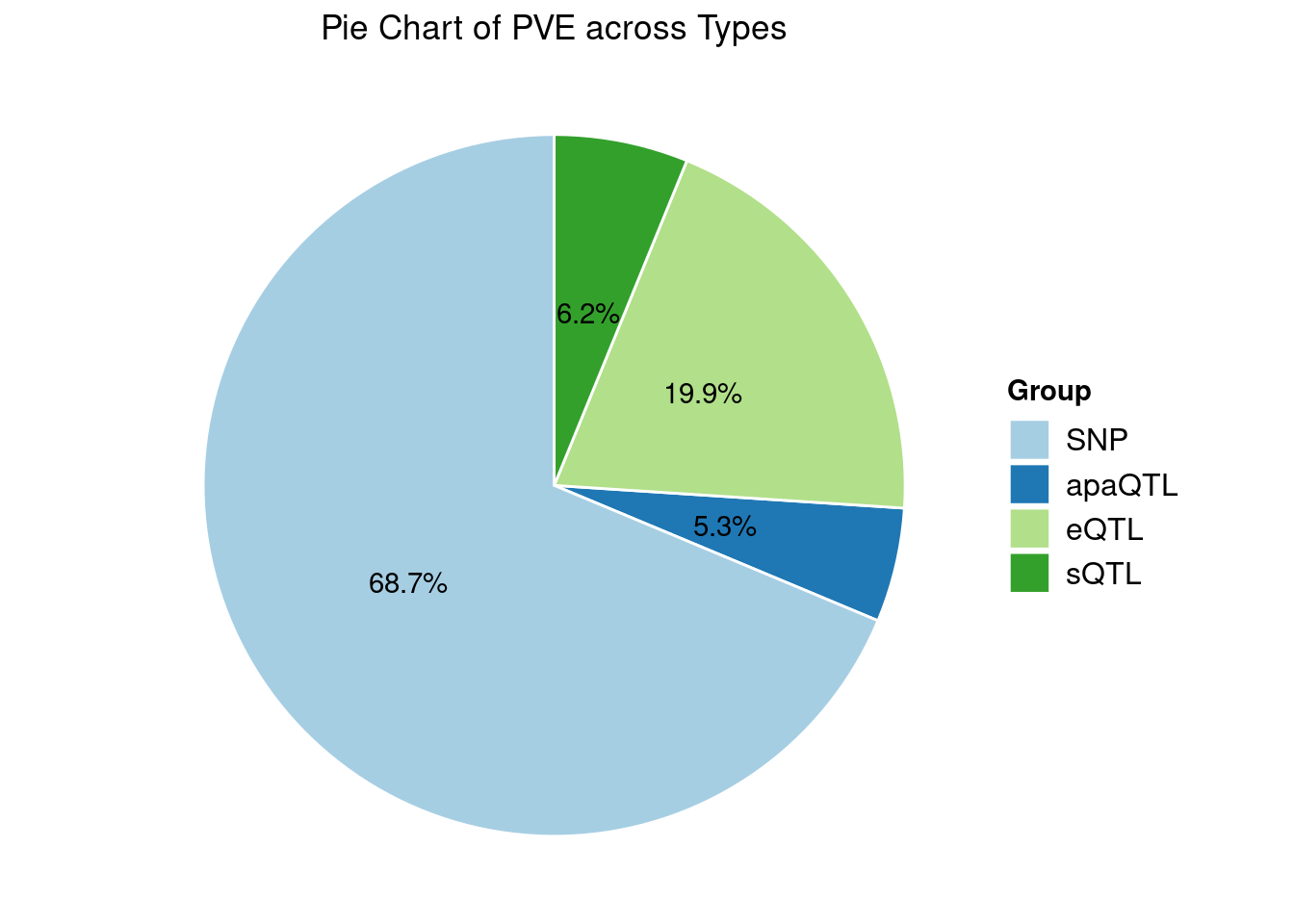
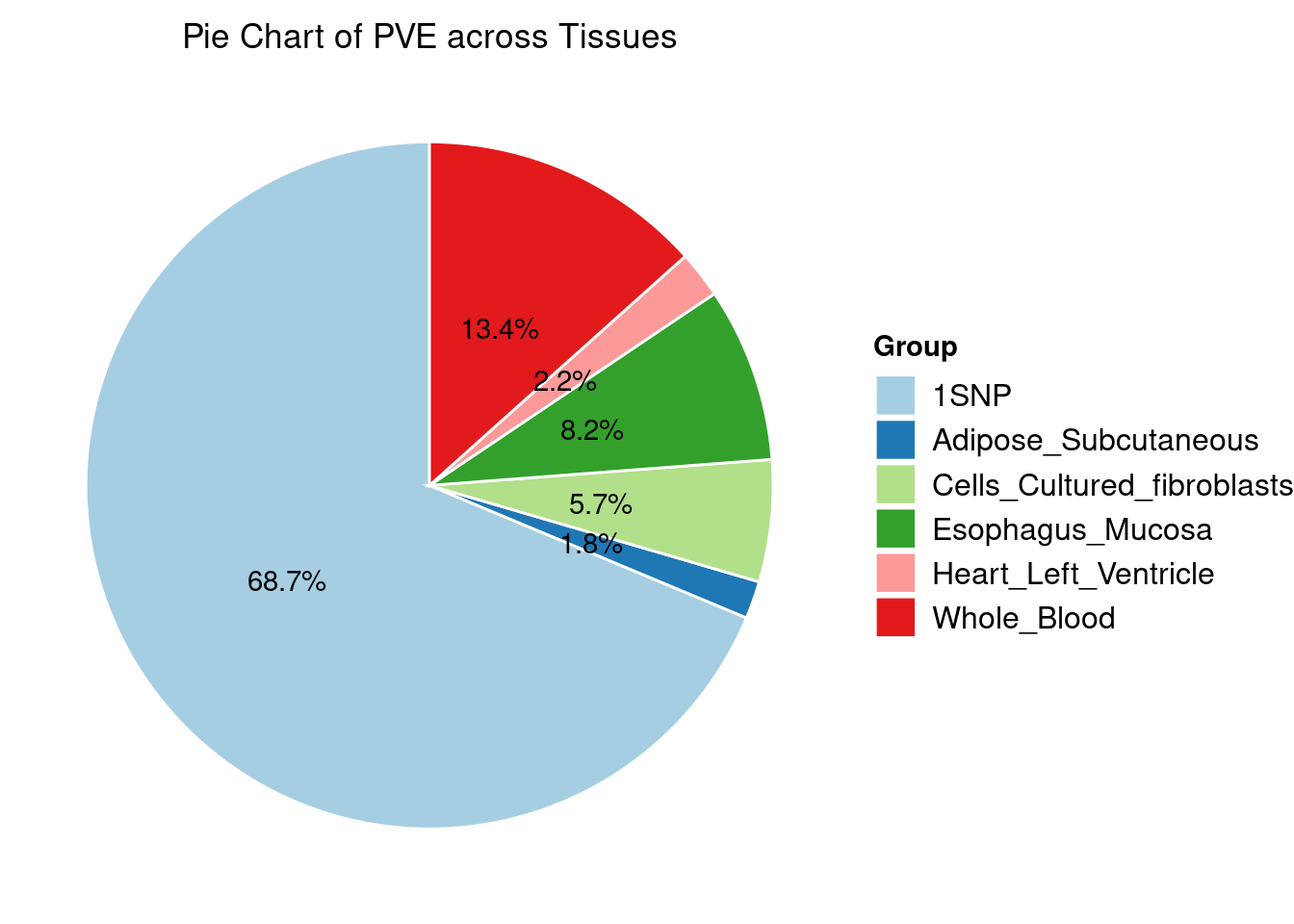
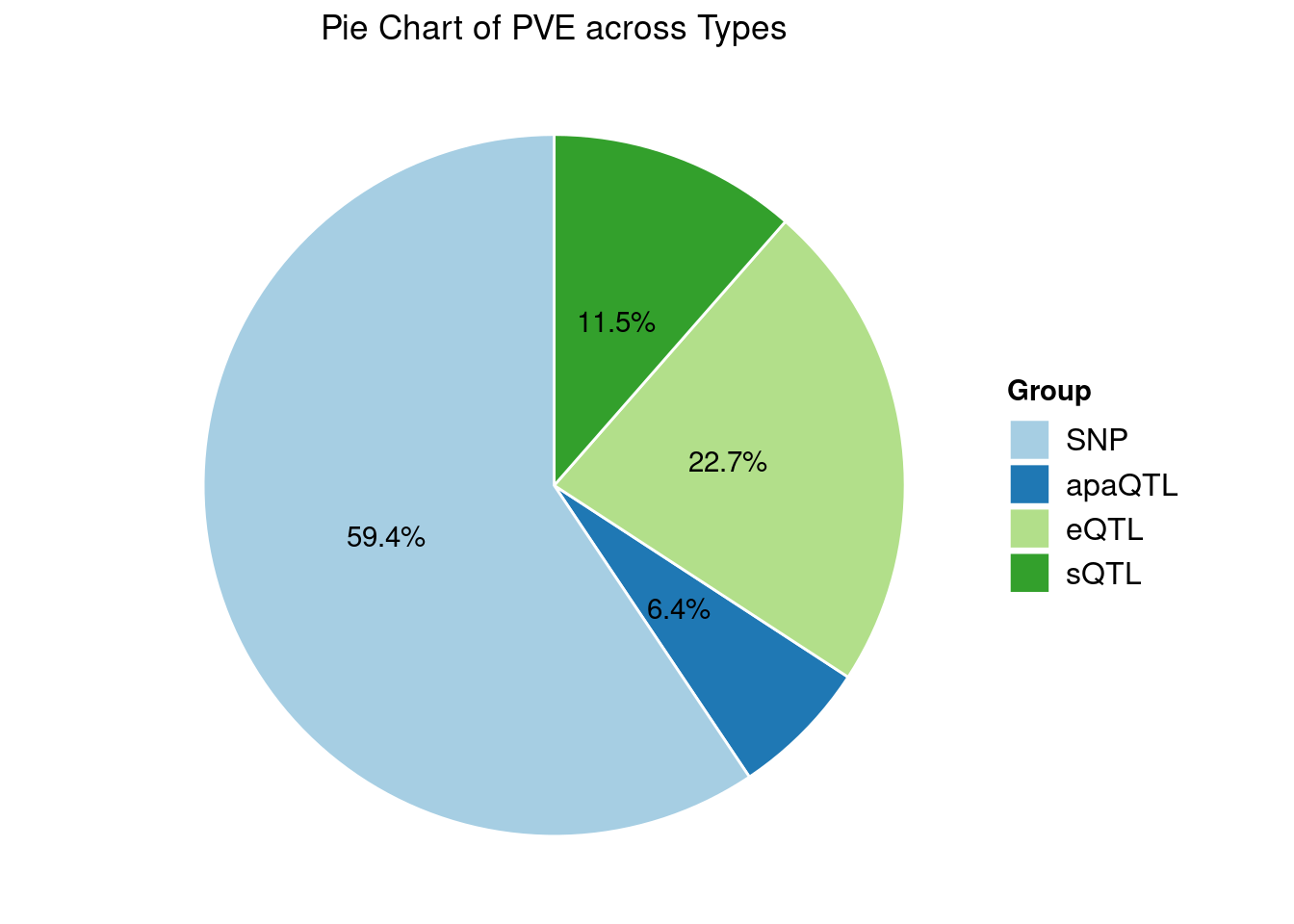
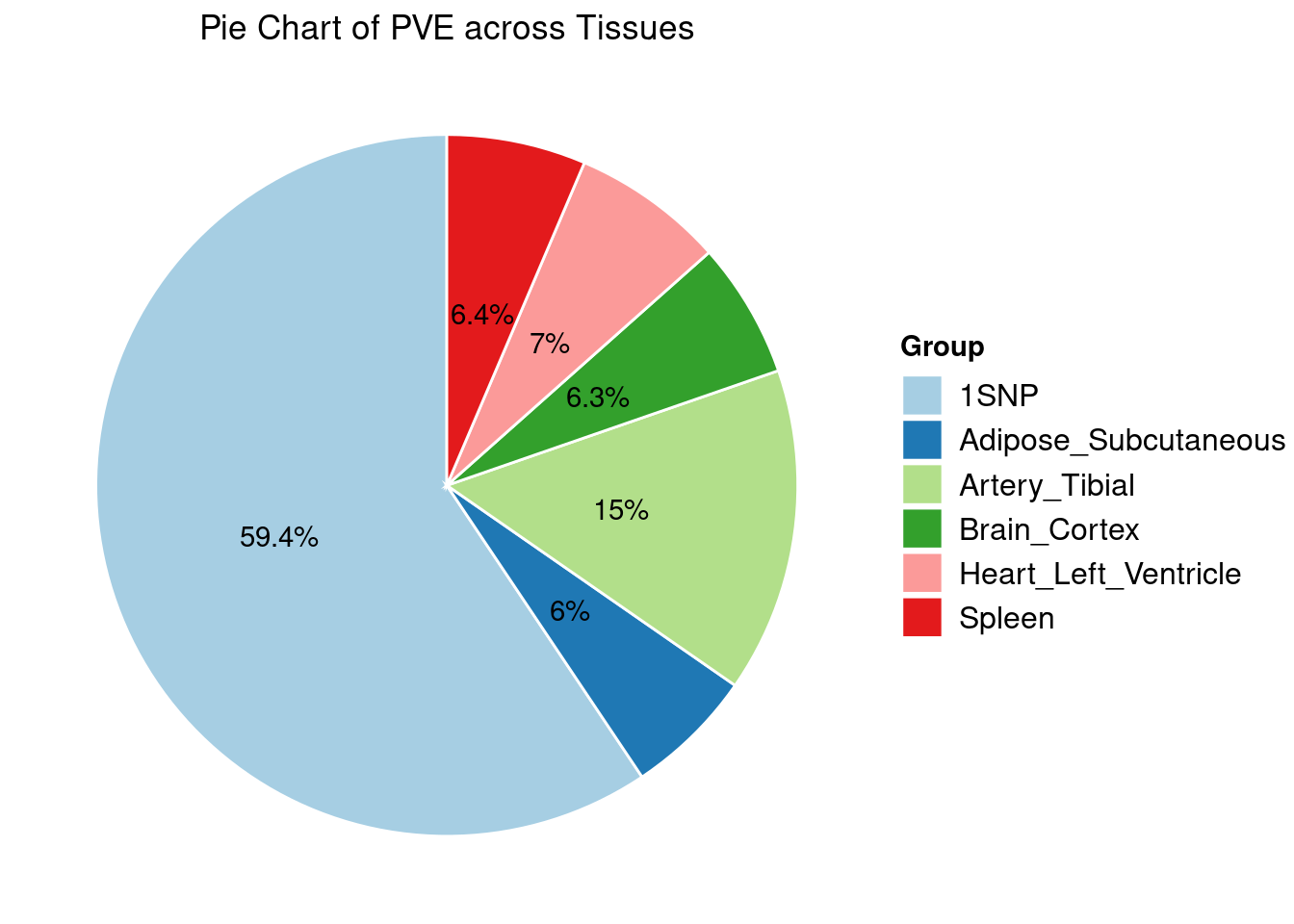
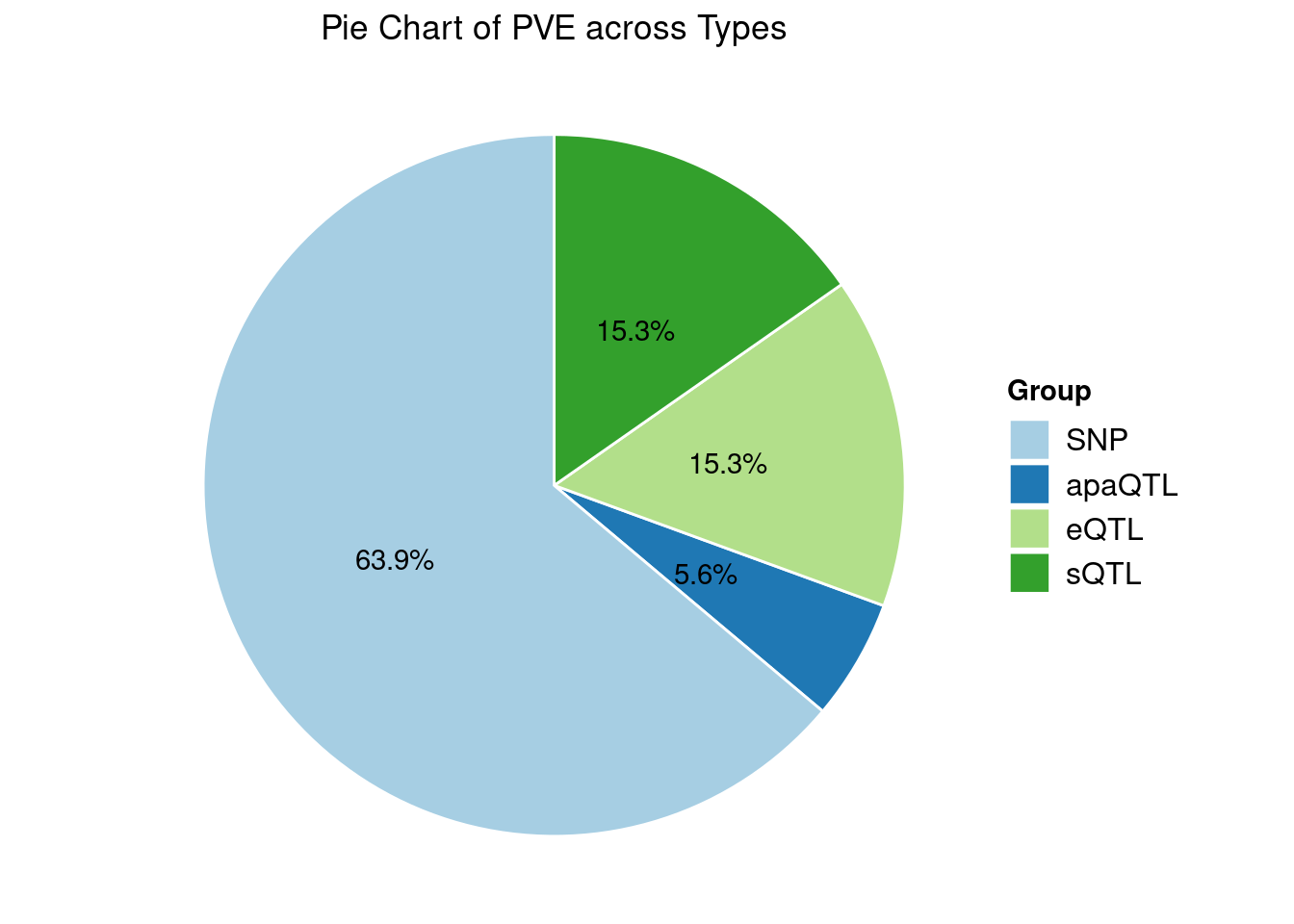
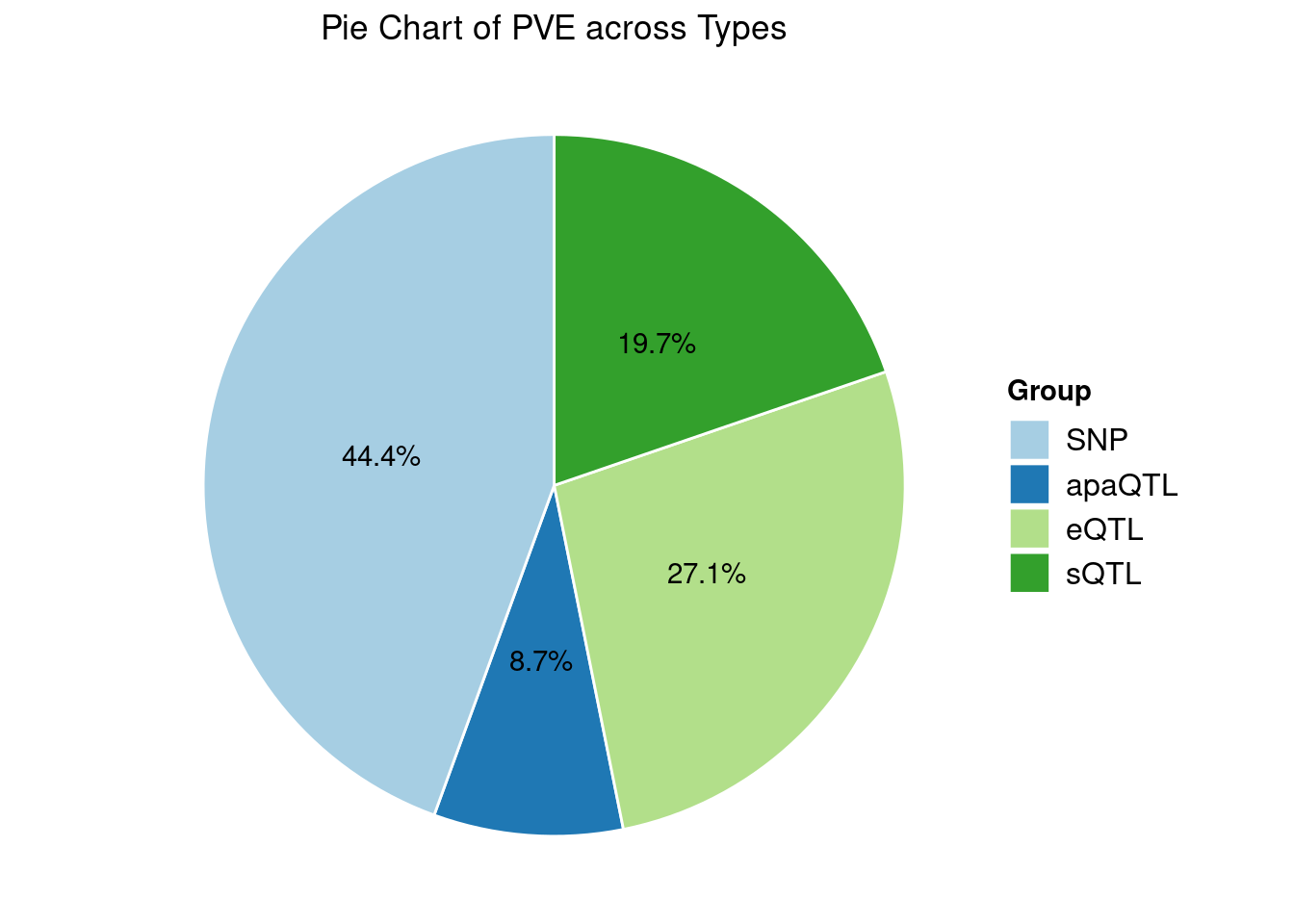
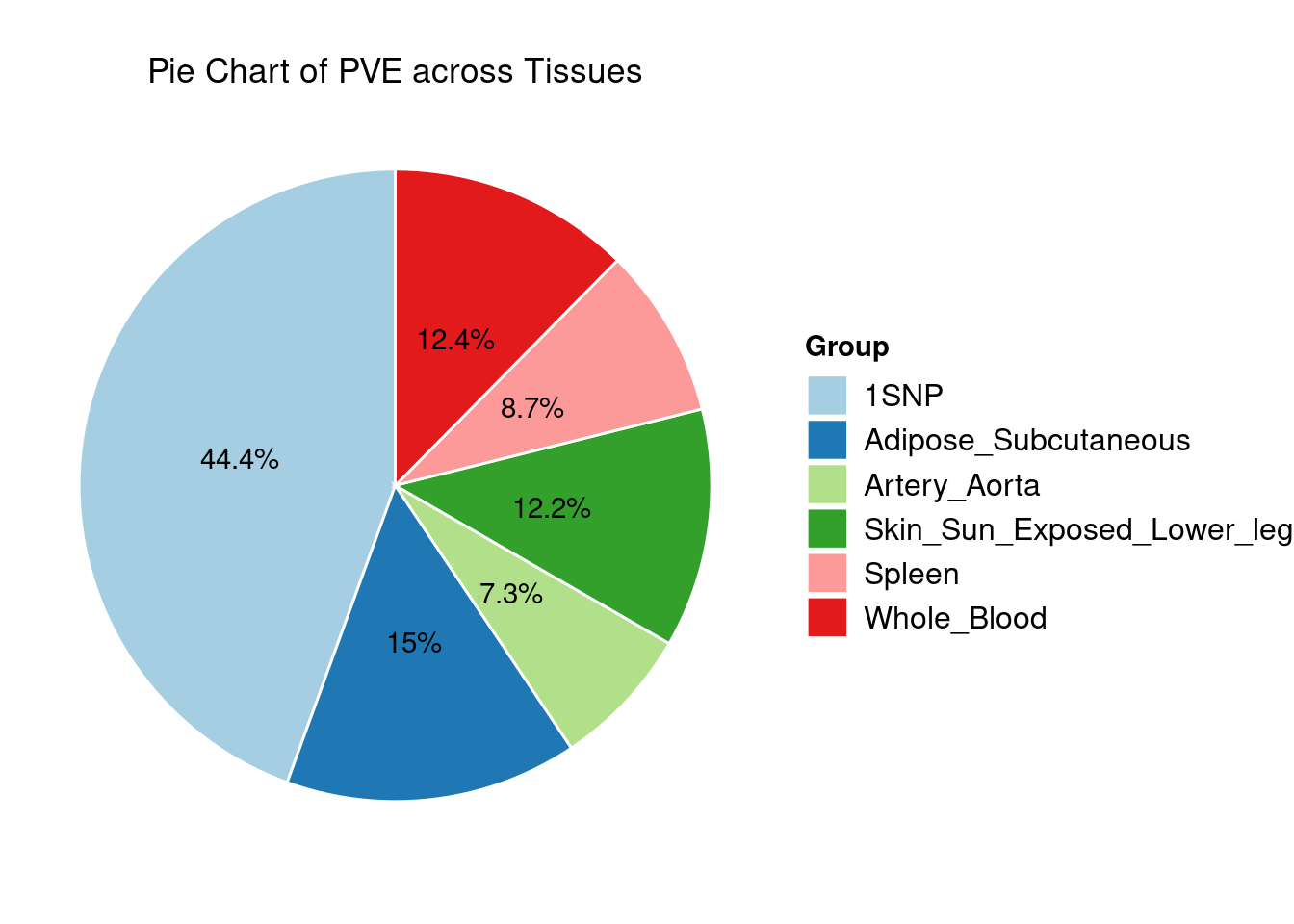
Heritability contribution by contexts: we aggregate the PVE values by omics and tissues, making it easier to understand the distribution of PVE across different genetic contexts.
Combined PIP by omics: we aggregate the Susie PIPs by omics
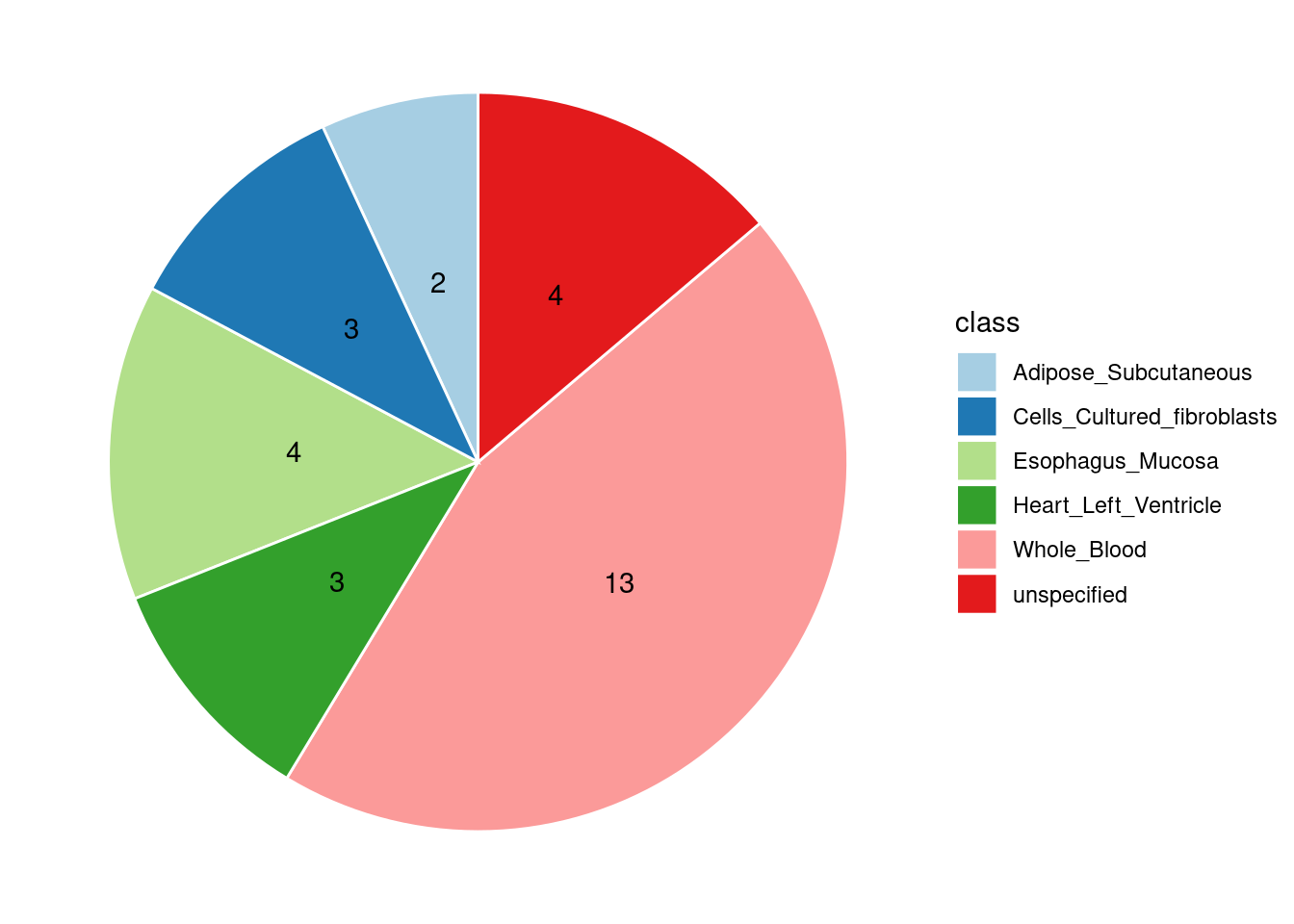
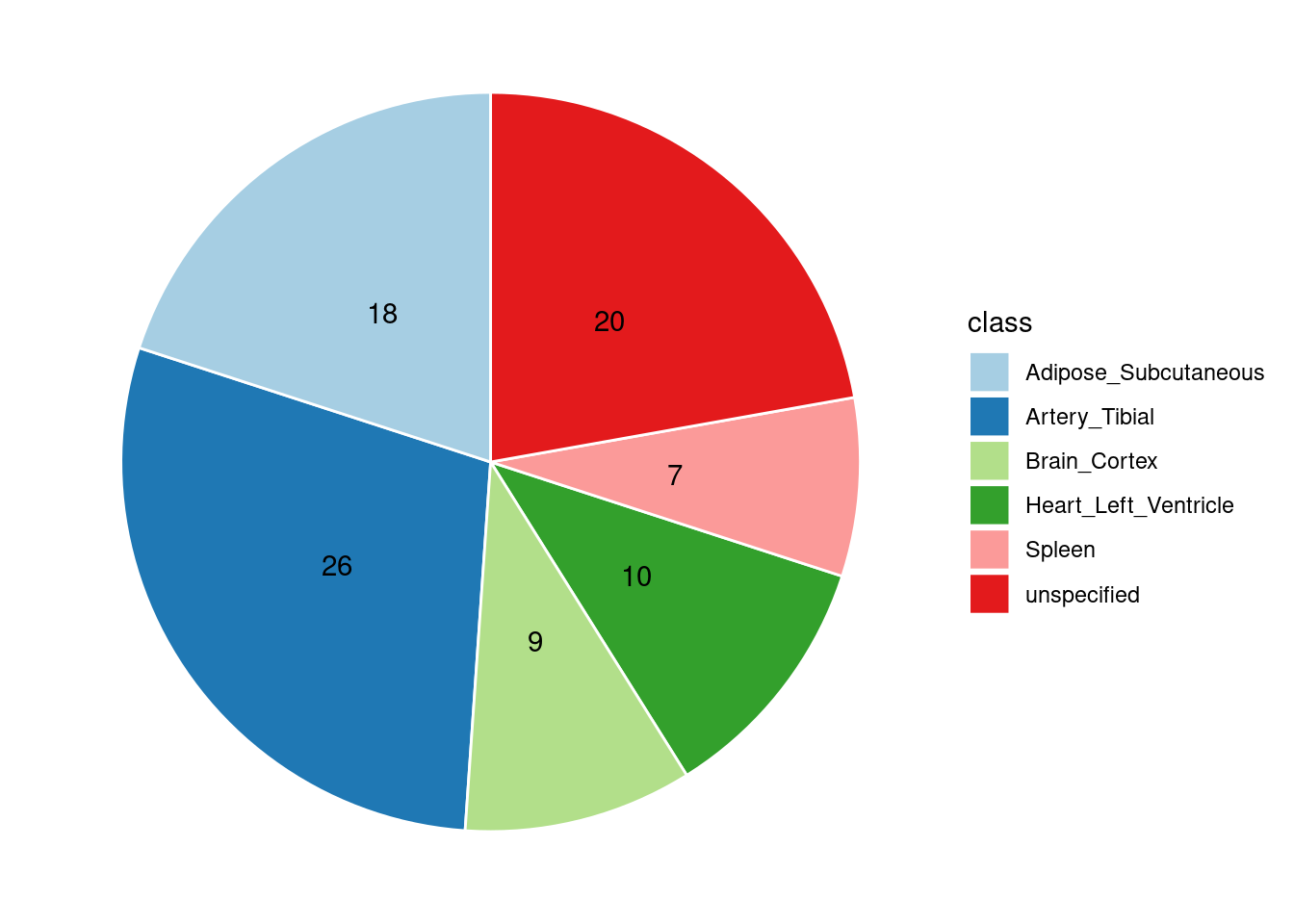
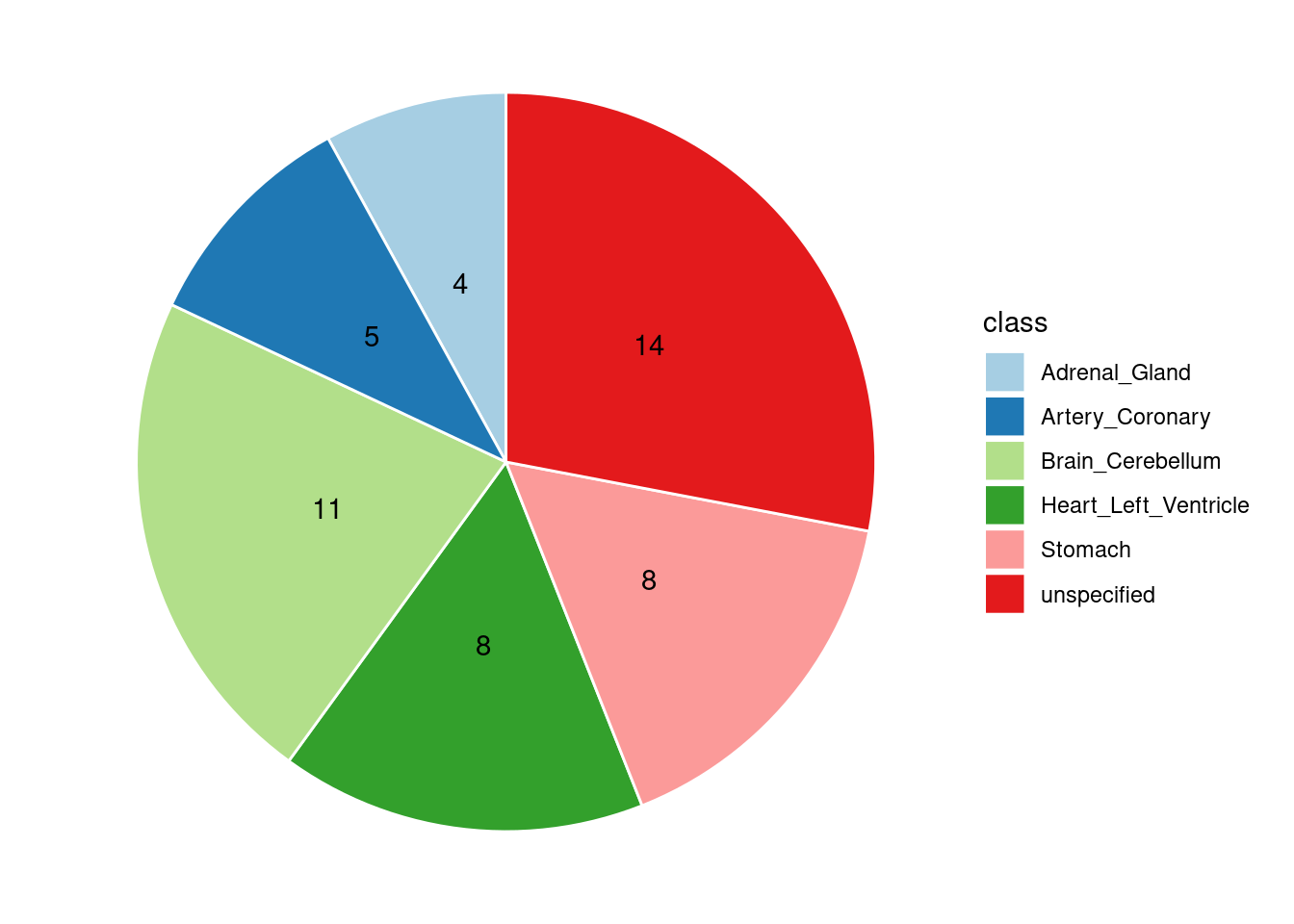
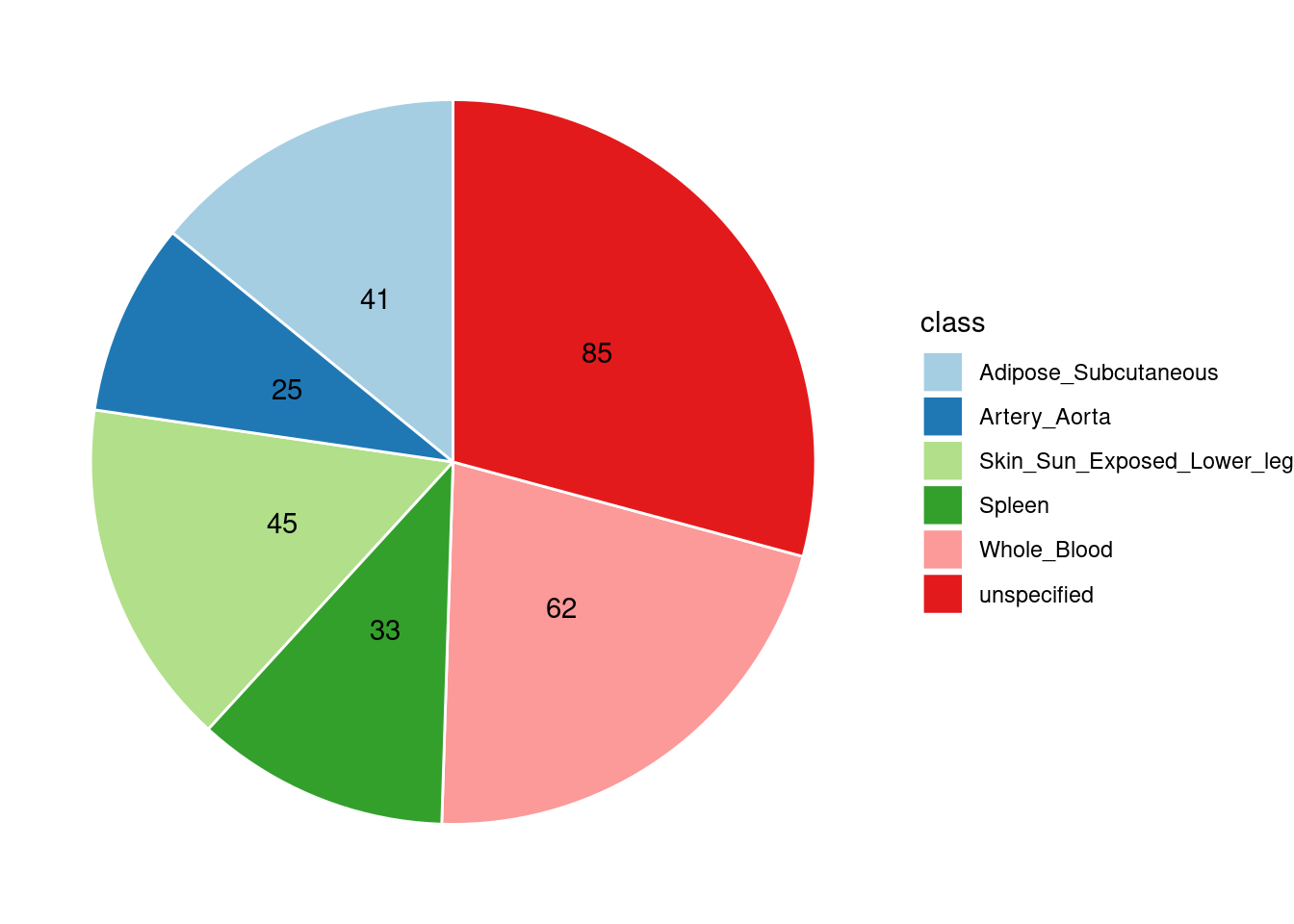
Combined PIP by contexts: we aggregate the Susie PIPs by tissues, making it easier to understand the distribution of PIP across different genetic contexts.
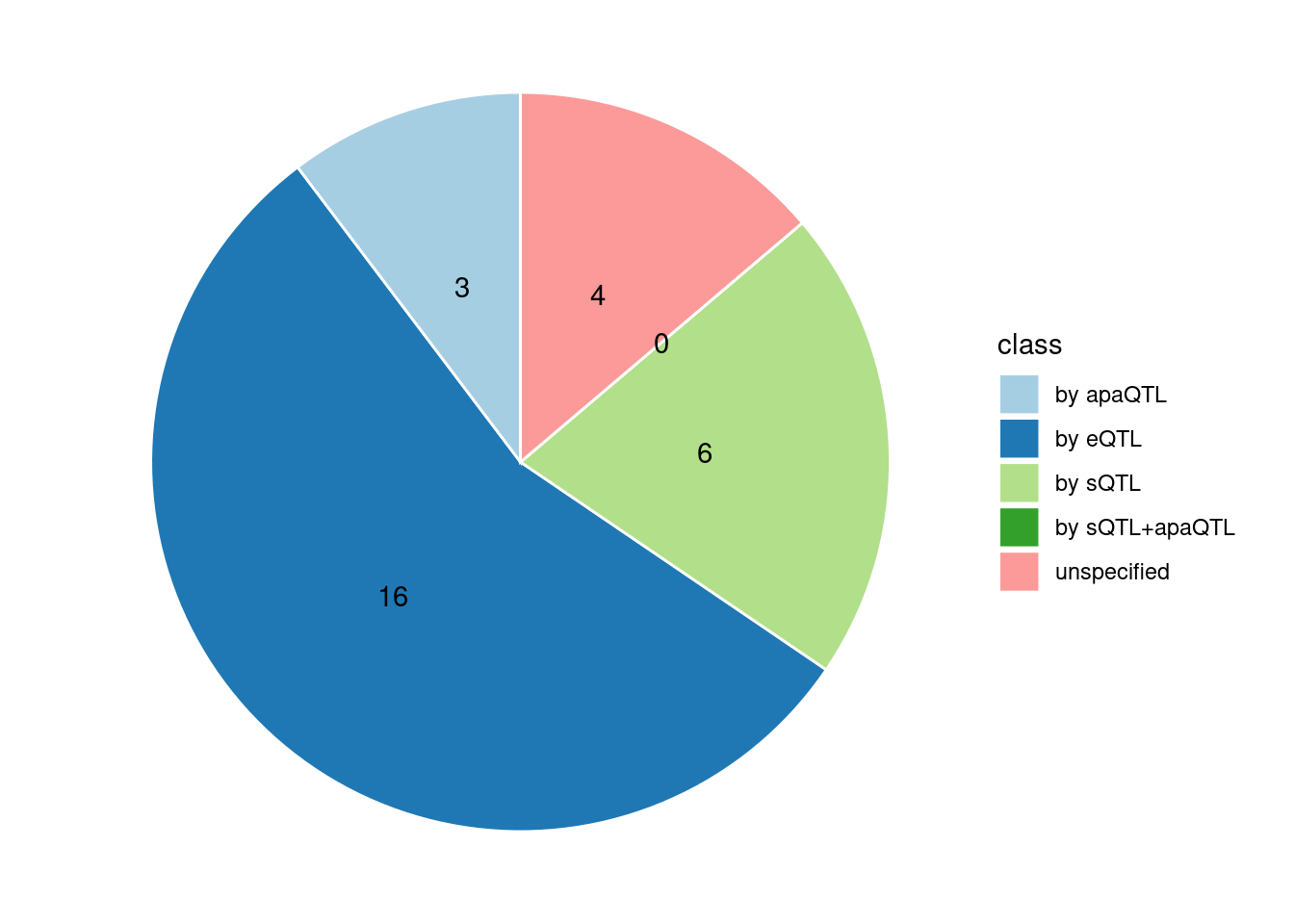
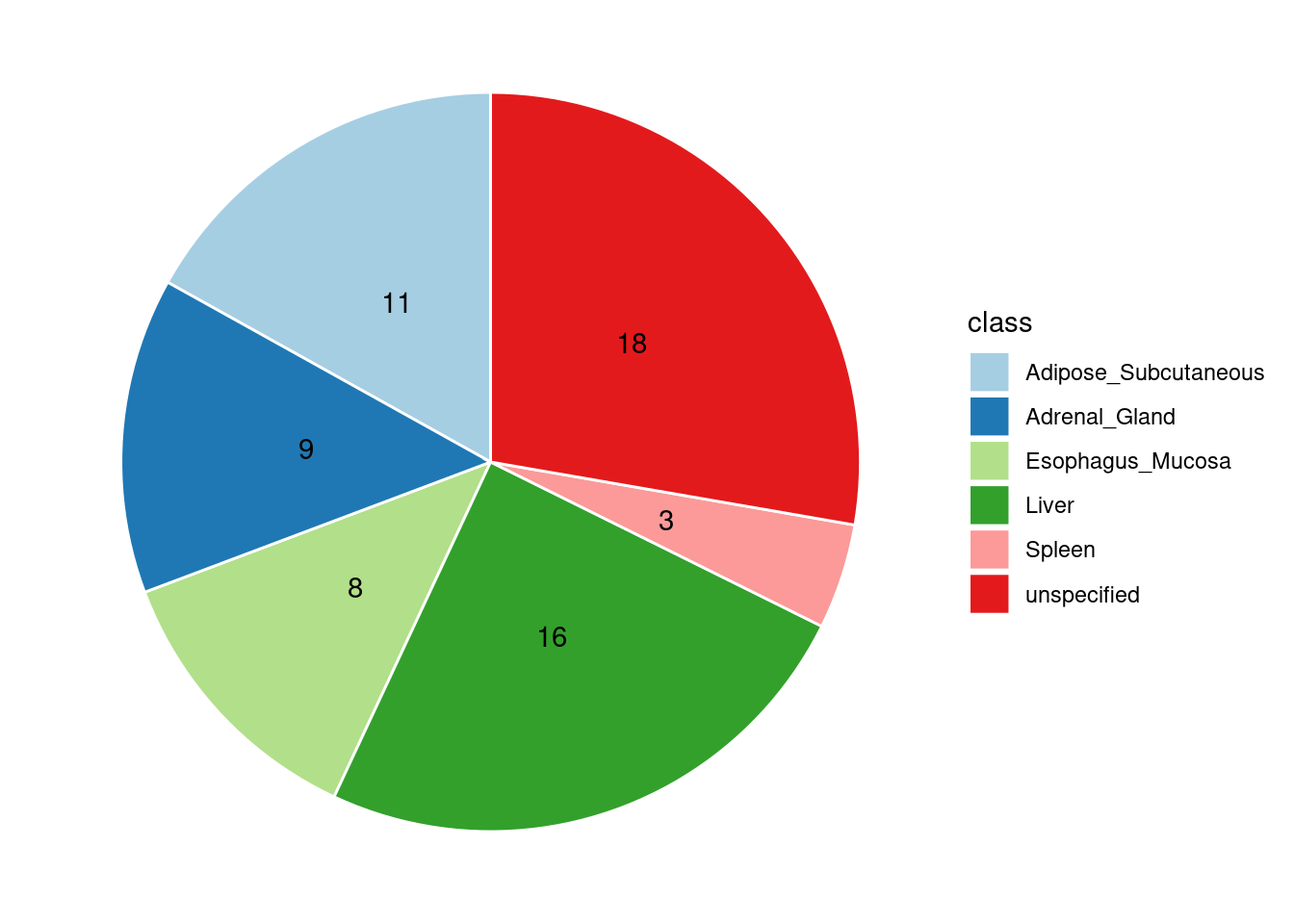
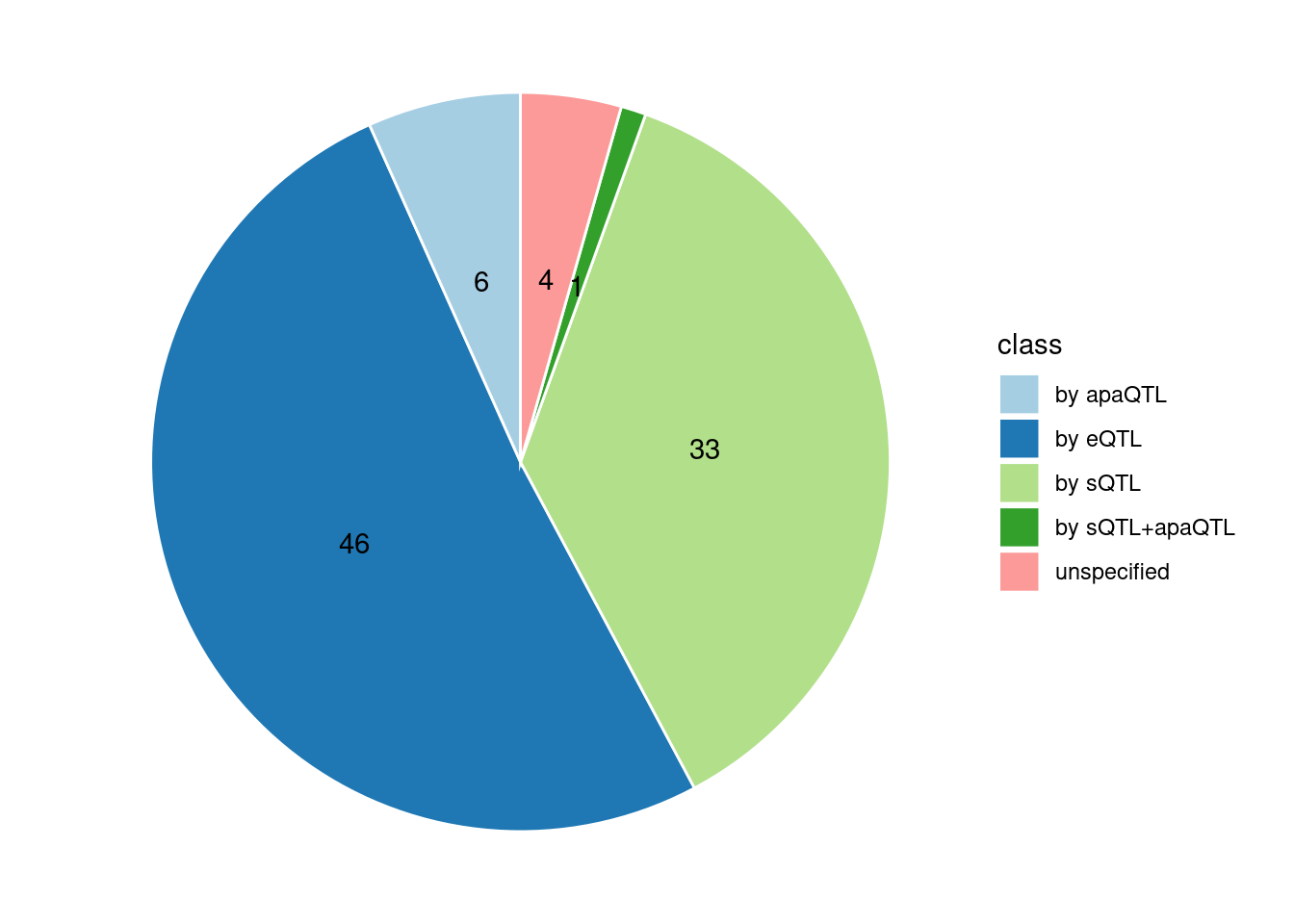
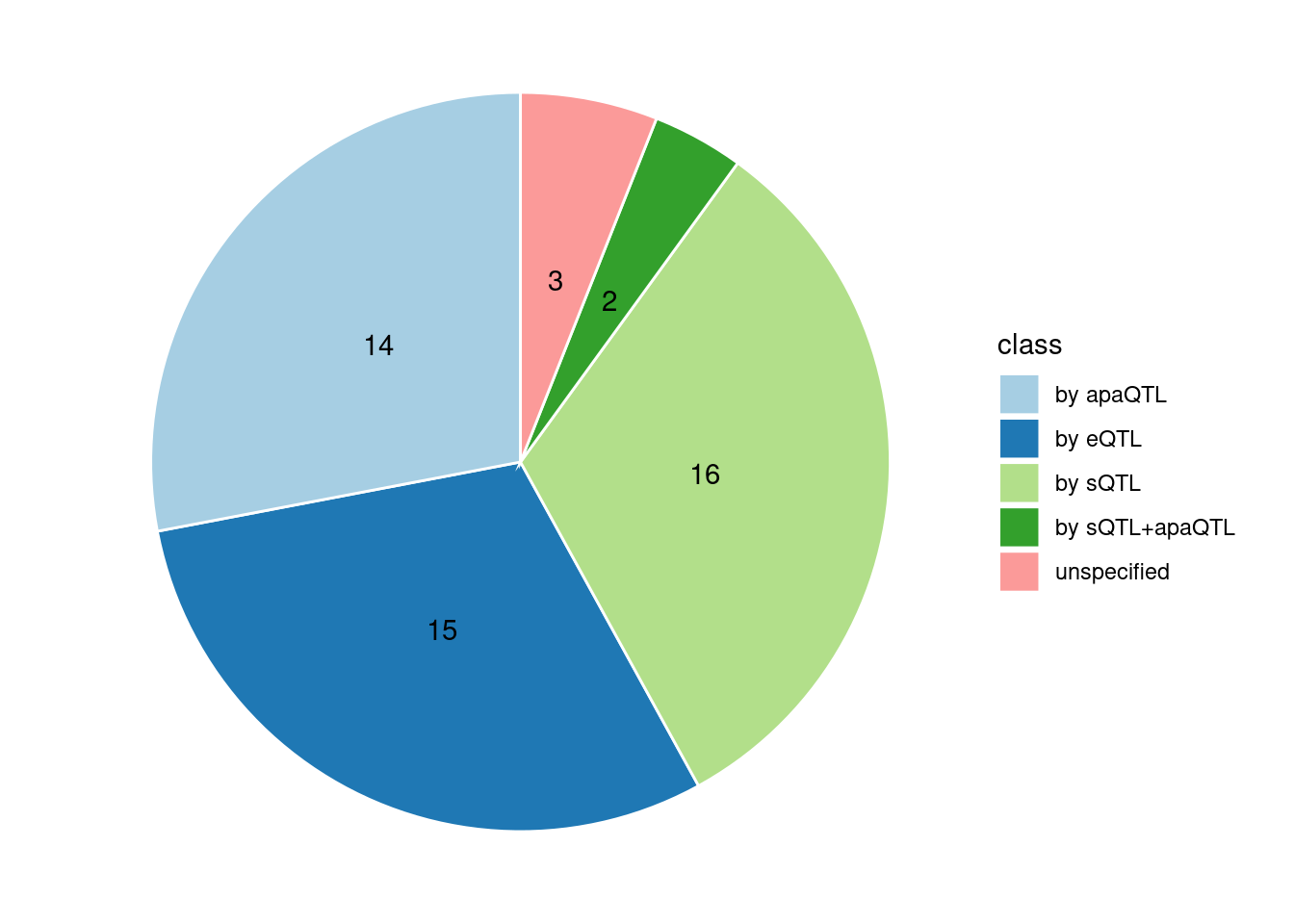
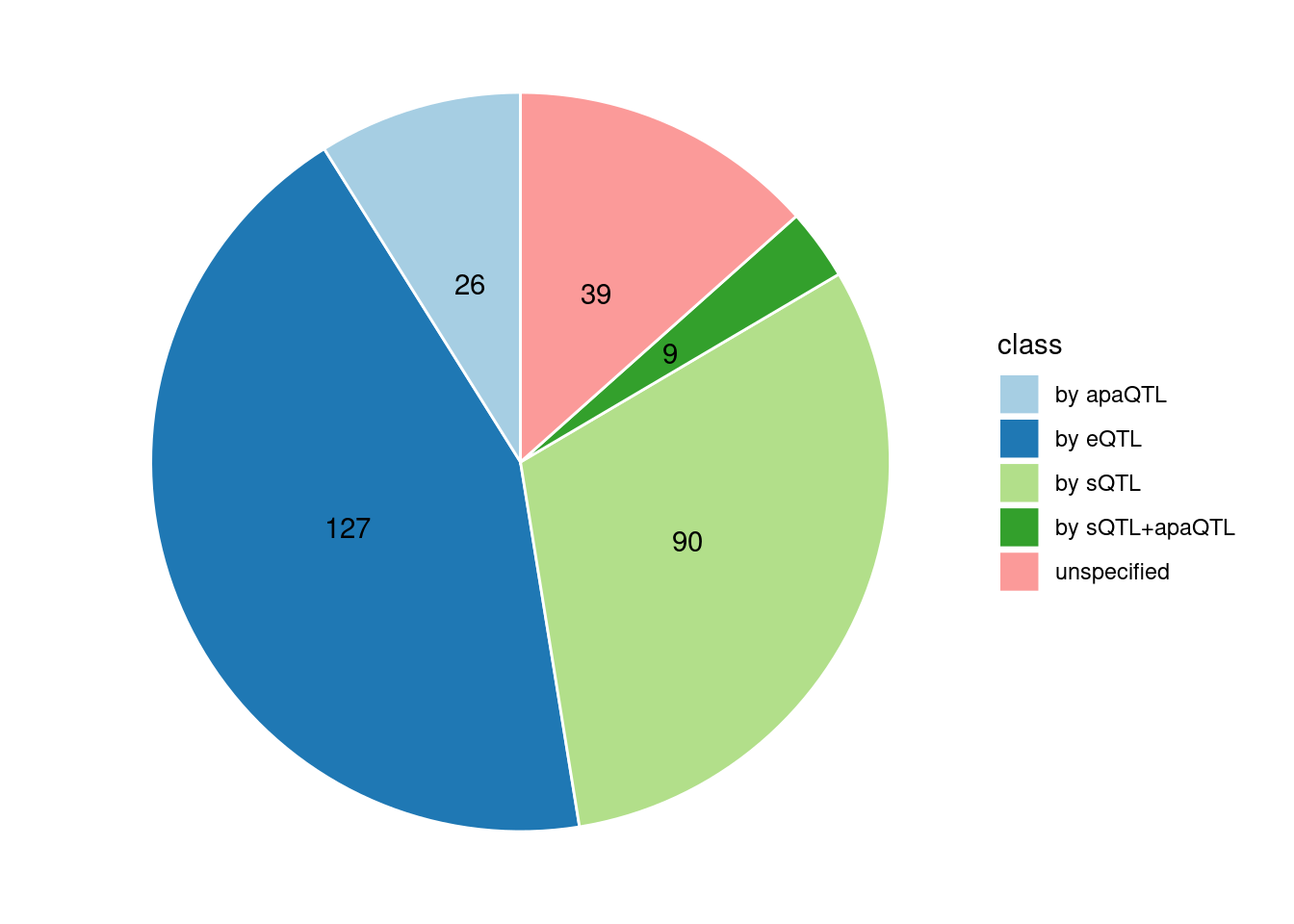
Specific molecular traits of top genes: we creates a pie chart to visualize the proportion of genes classified into different categories based on their PIPs contributed by each genetics contexts. The categories are based on the proportion of each QTL type relative to the combined PIP value:
- by eQTL: Number of genes where the ratio of eQTL to combined PIP is greater than 0.8.
- by sQTL: Number of genes where the ratio of sQTL to combined PIP is greater than 0.8.
- by apaQTL: Number of genes where the ratio of apaQTL to combined PIP is greater than 0.8.
- by sQTL+apaQTL: Number of genes where the combined ratio of apaQTL and sQTL to combined PIP is greater than 0.8, but neither apaQTL nor sQTL individually exceed 0.8.
- unspecified: Number of genes not classified into any of the above categories.
Comparing with earlier multi-group analysis results
We compared number of significant genes, overlapping genes. The earlier results are here: https://sq-96.github.io/multigroup_ctwas_analysis/multi_group_6traits_15weights_ukbb.html
Sigificant gene compare summary
| sig_gene_current | sig_gene_earlier | sig_gene_overlap | groupsize_eqtl_current | groupsize_eqtl_earlier | groupsize_sqtl_current | groupsize_sqtl_earlier | groupsize_apaqtl_current | groupsize_apaqtl_earlier | |
|---|---|---|---|---|---|---|---|---|---|
| ibd | 29 | 32 | 9 | 6034-5060-5880-5700-3931 | 9791-8784-9944-9984-8896 | 7198-5497-8911-6912-4261 | 24290-16109-28632-24379-19217 | 6789-4705-7036-5922-3955 | 1503-1973-1568-698-827 |
| ldl | 65 | 67 | 12 | 3194-5209-8951-4409-6977 | 8775-9749-10538-9433-10580 | 3194-5209-8951-4409-6977 | 18136-27105-30032-24941-25674 | 2700-4414-7076-3740-5950 | 1986-1578-636-575-1025 |
| sbp | 90 | 84 | 27 | 5903-5902-3412-3947-4095 | 10137 -10071-9201 -8977-9234 | 8224-8950-4089-4290-5206 | 27192 -28744-22553-19343-25955 | 6667-7076-3619-3970-4414 | 1980-1647-571-1003-703 |
| scz | 50 | 28 | 4 | 3936-3375-2540-4542-3386 | 8486-8397-8425-8675-8695 | 4270-4400-4247-5593-4960 | 18100-22295-24656-24890-22249 | 3951-3365-4225-4349-3729 | 1018-693-770-1056-1194 |
| wbc | 291 | 220 | 82 | 5078-5891-4790-6294-4088 | 8455-9589-9402-9943-8721 | 5530-8927-6490-9102-5185 | 15248-27095-25101-26876-24403 | 4700-7055-5281-7227-4409 | 1973-1479-1779-571-821 |
aFib
TO DO
IBD
Results from multi-group analysis
| Version | Author | Date |
|---|---|---|
| 9e83189 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 9e83189 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 9e83189 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 9e83189 | XSun | 2024-06-13 |
[1] "Esophagus_Mucosa" "Adipose_Subcutaneous"
[3] "Whole_Blood" "Heart_Left_Ventricle"
[5] "Cells_Cultured_fibroblasts"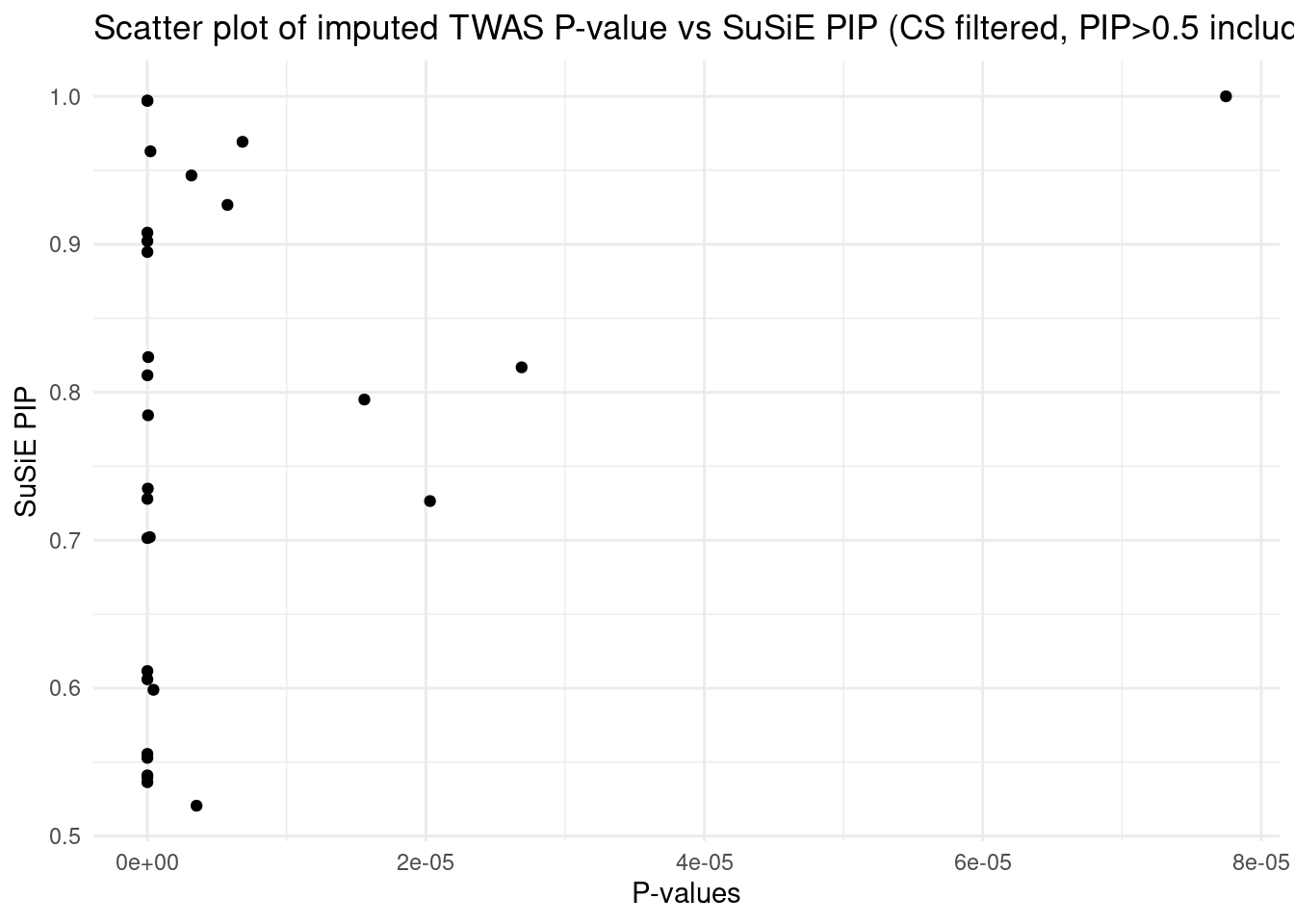
| Version | Author | Date |
|---|---|---|
| 9e83189 | XSun | 2024-06-13 |
Comparing with earlier weights
overlaps
[1] "current # of sig. genes = 29"[1] "earlier # of sig. genes = 32"[1] "# of overlap between them = 9"Why earlier genes were not reported in current settings
In the table below, NA means: current weights do not have this gene.
If combined_pip_current_weights >0.8, it means in
current setting, this gene is not included in CS.
combined_pip_current_weights < 0.8 means in current
setting, the gene is filtered out by either credible set or
combined_pip.
[1] "8 were not included (NAs) because they are not in the weight files"[1] "1 were not included (combined_pip > 0.8) because they are not in CS"[1] "14 were not included (combined_pip < 0.8) because of the low combined PIPs"Why current genes were not reported in earlier settings
In the table below, NA means: earlier weights do not have this gene.
If combined_pip_current_weights >0.8, it means in
earlier setting, this gene is not included in CS.
combined_pip_earlier_weights < 0.8 means in earlier
setting, the gene is filtered out by either credible set or
combined_pip.
[1] "3 were not included (NAs) because they are not in the weight files"[1] "0 were not included (combined_pip > 0.8) because they are not in CS"[1] "17 were not included (combined_pip < 0.8) because of the low combined PIPs"LDL
Results from multi-group analysis
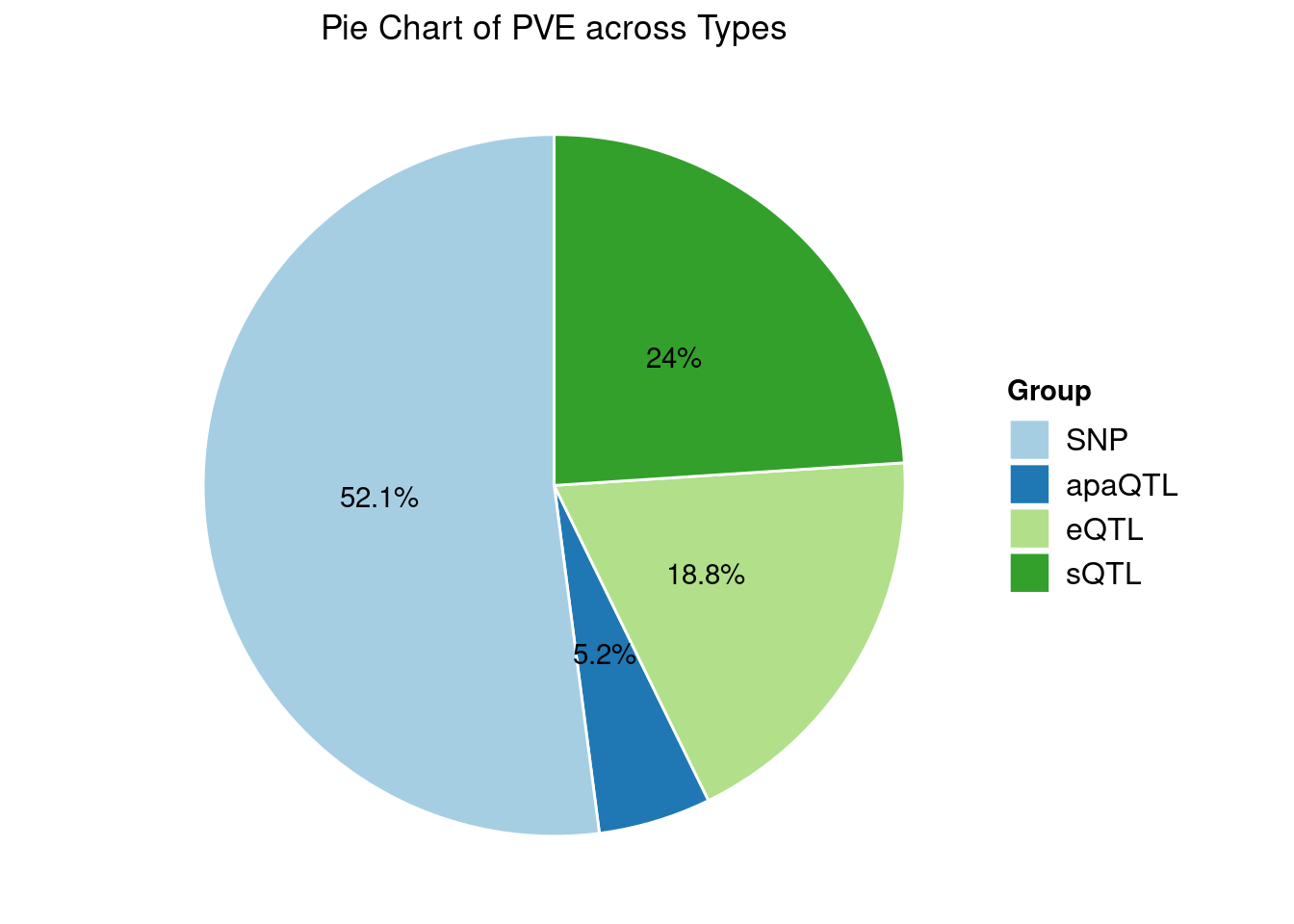
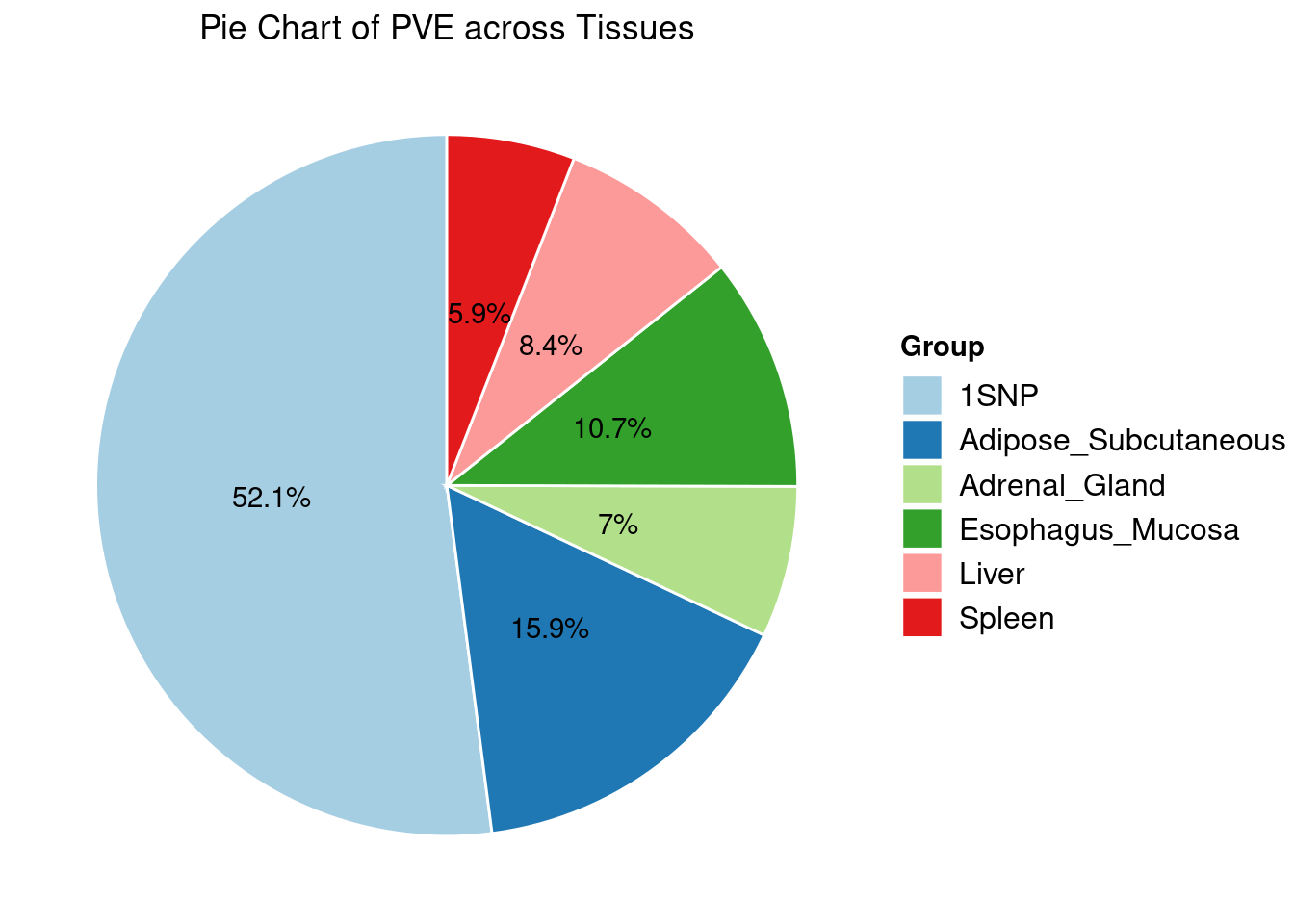
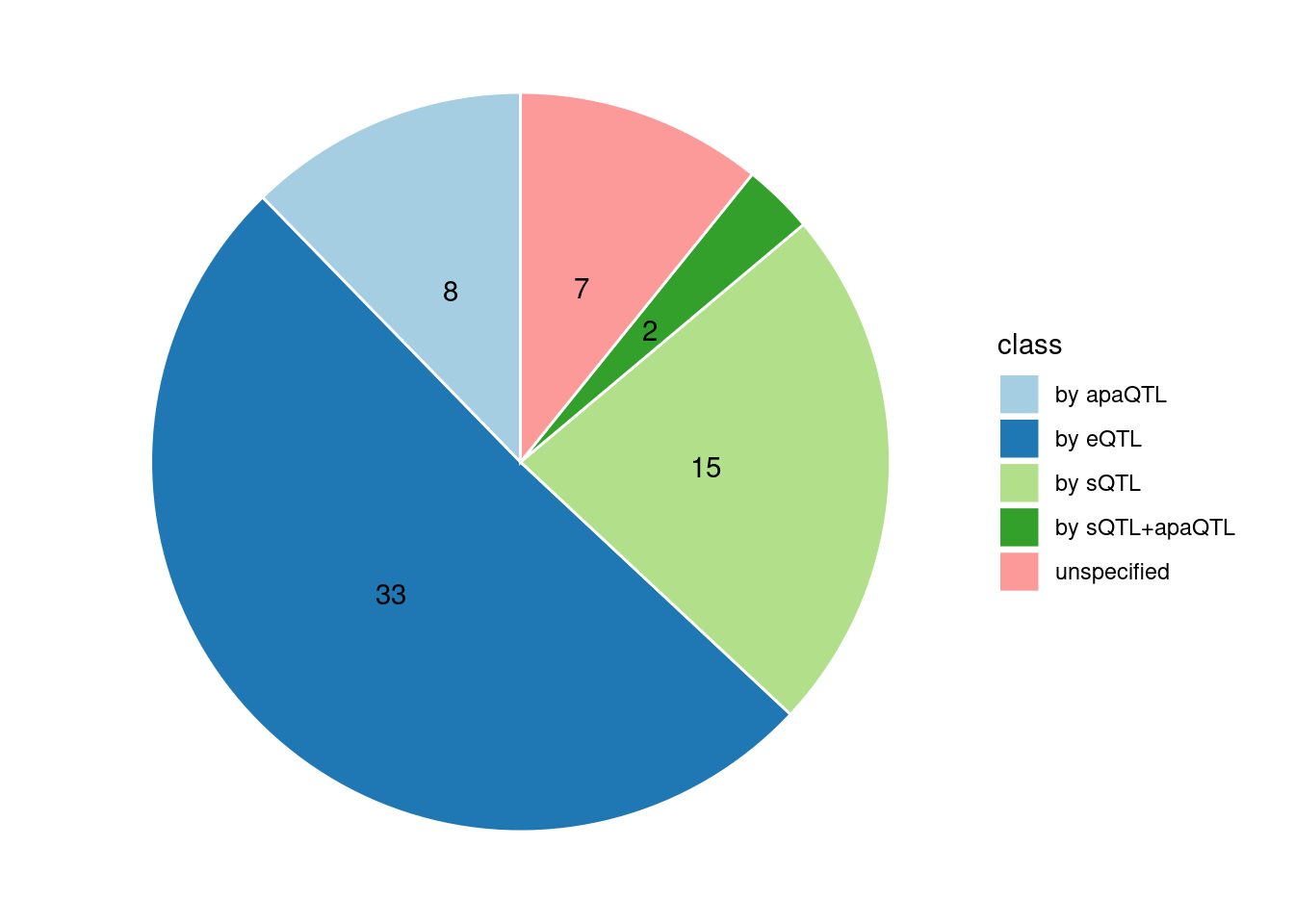
[1] "Esophagus_Mucosa" "Adipose_Subcutaneous" "Liver"
[4] "Adrenal_Gland" "Spleen" 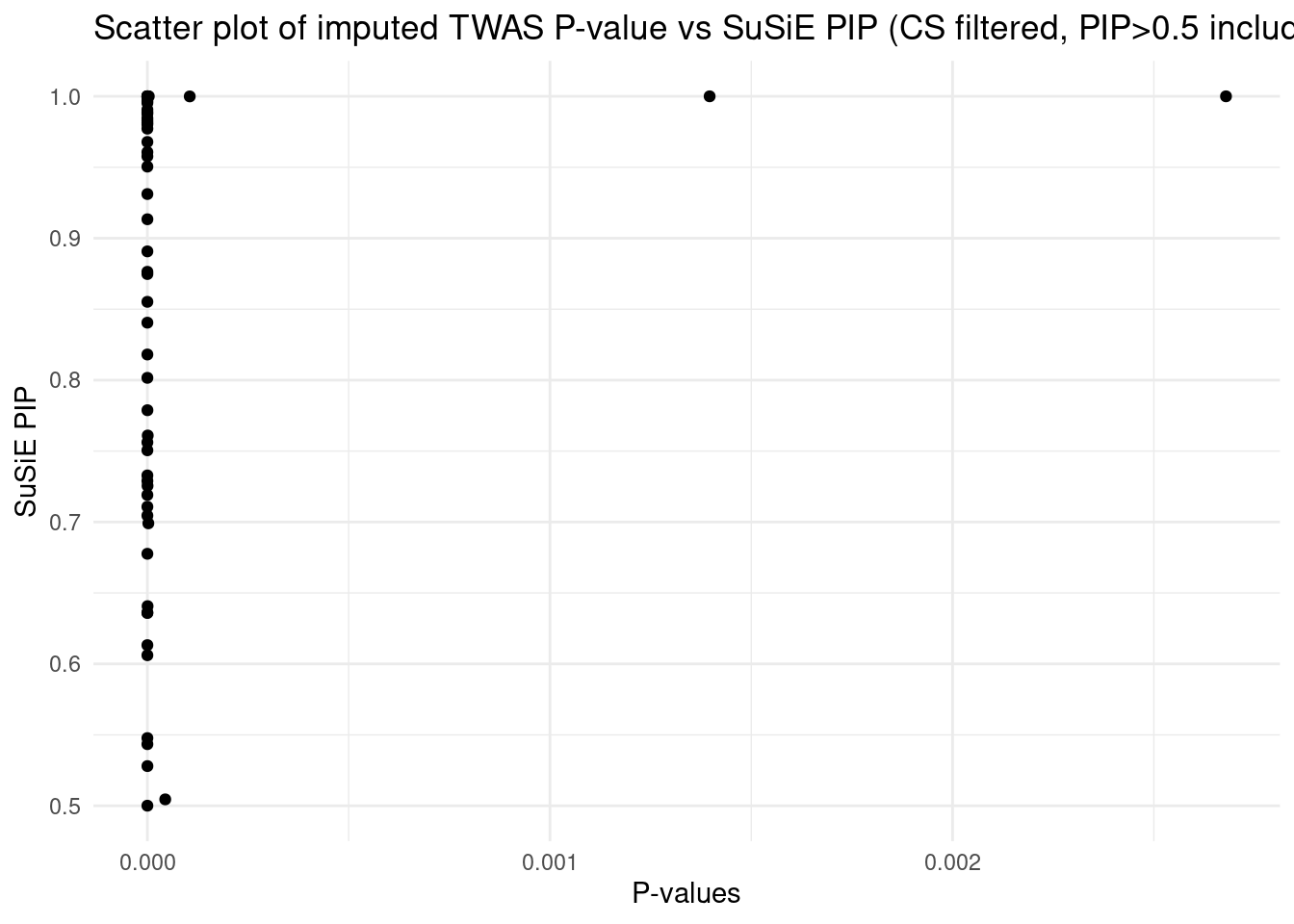
Comparing with earlier weights
overlaps
[1] "current # of sig. genes = 65"[1] "earlier # of sig. genes = 67"[1] "# of overlap between them = 12"Why earlier genes were not reported in current settings
In the table below, NA means: current weights do not have this gene.
If combined_pip_current_weights >0.8, it means in
current setting, this gene is not included in CS.
combined_pip_current_weights < 0.8 means in current
setting, the gene is filtered out by either credible set or
combined_pip.
[1] "23 were not included (NAs) because they are not in the weight files"[1] "2 were not included (combined_pip > 0.8) because they are not in CS"[1] "30 were not included (combined_pip < 0.8) because of the low combined PIPs"Why current genes were not reported in earlier settings
In the table below, NA means: earlier weights do not have this gene.
If combined_pip_current_weights >0.8, it means in
earlier setting, this gene is not included in CS.
combined_pip_earlier_weights < 0.8 means in earlier
setting, the gene is filtered out by either credible set or
combined_pip.
[1] "7 were not included (NAs) because they are not in the weight files"[1] "0 were not included (combined_pip > 0.8) because they are not in CS"[1] "46 were not included (combined_pip < 0.8) because of the low combined PIPs"SBP
Results from multi-group analysis
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
[1] "Artery_Tibial" "Heart_Left_Ventricle" "Spleen"
[4] "Adipose_Subcutaneous" "Brain_Cortex" 
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
Comparing with earlier weights
overlaps
[1] "current # of sig. genes = 90"[1] "earlier # of sig. genes = 84"[1] "# of overlap between them = 27"Why earlier genes were not reported in current settings
In the table below, NA means: current weights do not have this gene.
If combined_pip_current_weights >0.8, it means in
current setting, this gene is not included in CS.
combined_pip_current_weights < 0.8 means in current
setting, the gene is filtered out by either credible set or
combined_pip.
[1] "17 were not included (NAs) because they are not in the weight files"[1] "2 were not included (combined_pip > 0.8) because they are not in CS"[1] "38 were not included (combined_pip < 0.8) because of the low combined PIPs"Why current genes were not reported in earlier settings
In the table below, NA means: earlier weights do not have this gene.
If combined_pip_current_weights >0.8, it means in
earlier setting, this gene is not included in CS.
combined_pip_earlier_weights < 0.8 means in earlier
setting, the gene is filtered out by either credible set or
combined_pip.
[1] "10 were not included (NAs) because they are not in the weight files"[1] "2 were not included (combined_pip > 0.8) because they are not in CS"[1] "51 were not included (combined_pip < 0.8) because of the low combined PIPs"SCZ
Results from multi-group analysis
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
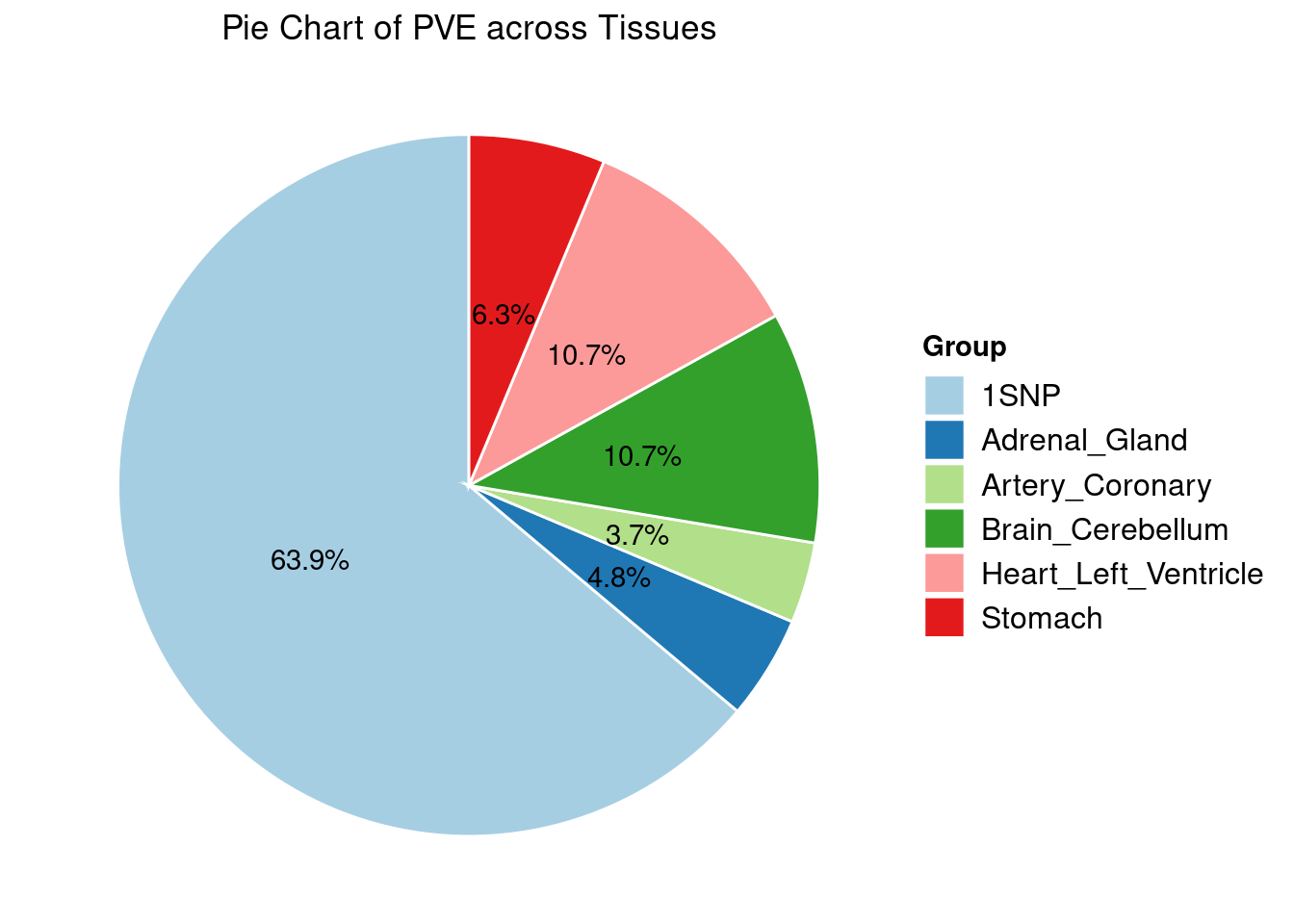
[1] "Heart_Left_Ventricle" "Adrenal_Gland" "Brain_Cerebellum"
[4] "Stomach" "Artery_Coronary" 
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
Comparing with earlier weights
overlaps
[1] "current # of sig. genes = 50"[1] "earlier # of sig. genes = 28"[1] "# of overlap between them = 4"Why earlier genes were not reported in current settings
In the table below, NA means: current weights do not have this gene.
If combined_pip_current_weights >0.8, it means in
current setting, this gene is not included in CS.
combined_pip_current_weights < 0.8 means in current
setting, the gene is filtered out by either credible set or
combined_pip.
[1] "13 were not included (NAs) because they are not in the weight files"[1] "1 were not included (combined_pip > 0.8) because they are not in CS"[1] "10 were not included (combined_pip < 0.8) because of the low combined PIPs"Why current genes were not reported in earlier settings
In the table below, NA means: earlier weights do not have this gene.
If combined_pip_current_weights >0.8, it means in
earlier setting, this gene is not included in CS.
combined_pip_earlier_weights < 0.8 means in earlier
setting, the gene is filtered out by either credible set or
combined_pip.
[1] "8 were not included (NAs) because they are not in the weight files"[1] "3 were not included (combined_pip > 0.8) because they are not in CS"[1] "35 were not included (combined_pip < 0.8) because of the low combined PIPs"WBC
Results from multi-group analysis
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
[1] "Whole_Blood" "Skin_Sun_Exposed_Lower_leg"
[3] "Adipose_Subcutaneous" "Artery_Aorta"
[5] "Spleen" 
| Version | Author | Date |
|---|---|---|
| 92a8e35 | XSun | 2024-06-13 |
Comparing with earlier weights
overlaps
[1] "current # of sig. genes = 291"[1] "earlier # of sig. genes = 220"[1] "# of overlap between them = 82"Why earlier genes were not reported in current settings
In the table below, NA means: current weights do not have this gene.
If combined_pip_current_weights >0.8, it means in
current setting, this gene is not included in CS.
combined_pip_current_weights < 0.8 means in current
setting, the gene is filtered out by either credible set or
combined_pip.
[1] "49 were not included (NAs) because they are not in the weight files"[1] "5 were not included (combined_pip > 0.8) because they are not in CS"[1] "84 were not included (combined_pip < 0.8) because of the low combined PIPs"Why current genes were not reported in earlier settings
In the table below, NA means: earlier weights do not have this gene.
If combined_pip_current_weights >0.8, it means in
earlier setting, this gene is not included in CS.
combined_pip_earlier_weights < 0.8 means in earlier
setting, the gene is filtered out by either credible set or
combined_pip.
[1] "26 were not included (NAs) because they are not in the weight files"[1] "13 were not included (combined_pip > 0.8) because they are not in CS"[1] "170 were not included (combined_pip < 0.8) because of the low combined PIPs"
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] gridExtra_2.3 RColorBrewer_1.1-3 forcats_0.5.1 stringr_1.5.1
[5] dplyr_1.1.4 purrr_1.0.2 readr_2.1.2 tidyr_1.3.0
[9] tibble_3.2.1 ggplot2_3.5.1 tidyverse_1.3.1 data.table_1.14.2
[13] ctwas_0.2.30
loaded via a namespace (and not attached):
[1] readxl_1.4.0 backports_1.4.1
[3] workflowr_1.7.0 BiocFileCache_2.4.0
[5] plyr_1.8.7 lazyeval_0.2.2
[7] crosstalk_1.2.0 BiocParallel_1.30.3
[9] GenomeInfoDb_1.39.9 LDlinkR_1.2.3
[11] digest_0.6.29 foreach_1.5.2
[13] ensembldb_2.20.2 htmltools_0.5.2
[15] fansi_1.0.3 magrittr_2.0.3
[17] memoise_2.0.1 doParallel_1.0.17
[19] tzdb_0.4.0 Biostrings_2.64.0
[21] modelr_0.1.8 matrixStats_0.62.0
[23] locuszoomr_0.2.1 prettyunits_1.1.1
[25] colorspace_2.0-3 blob_1.2.3
[27] rvest_1.0.2 rappdirs_0.3.3
[29] ggrepel_0.9.1 haven_2.5.0
[31] xfun_0.41 crayon_1.5.1
[33] RCurl_1.98-1.7 jsonlite_1.8.0
[35] zoo_1.8-10 iterators_1.0.14
[37] glue_1.6.2 gtable_0.3.0
[39] zlibbioc_1.42.0 XVector_0.36.0
[41] DelayedArray_0.22.0 BiocGenerics_0.42.0
[43] scales_1.3.0 DBI_1.2.2
[45] Rcpp_1.0.8.3 viridisLite_0.4.0
[47] progress_1.2.2 bit_4.0.4
[49] DT_0.22 stats4_4.2.0
[51] htmlwidgets_1.5.4 httr_1.4.3
[53] ellipsis_0.3.2 pkgconfig_2.0.3
[55] XML_3.99-0.14 farver_2.1.0
[57] sass_0.4.1 dbplyr_2.1.1
[59] utf8_1.2.2 tidyselect_1.2.0
[61] labeling_0.4.2 rlang_1.1.2
[63] later_1.3.0 AnnotationDbi_1.58.0
[65] munsell_0.5.0 pgenlibr_0.3.3
[67] cellranger_1.1.0 tools_4.2.0
[69] cachem_1.0.6 cli_3.6.1
[71] generics_0.1.2 RSQLite_2.3.1
[73] broom_0.8.0 evaluate_0.15
[75] fastmap_1.1.0 yaml_2.3.5
[77] knitr_1.39 bit64_4.0.5
[79] fs_1.5.2 KEGGREST_1.36.3
[81] AnnotationFilter_1.20.0 whisker_0.4
[83] xml2_1.3.3 biomaRt_2.54.1
[85] compiler_4.2.0 rstudioapi_0.13
[87] plotly_4.10.0 filelock_1.0.2
[89] curl_4.3.2 png_0.1-7
[91] reprex_2.0.1 bslib_0.3.1
[93] stringi_1.7.6 highr_0.9
[95] GenomicFeatures_1.48.3 lattice_0.20-45
[97] ProtGenerics_1.28.0 Matrix_1.5-3
[99] vctrs_0.6.5 pillar_1.9.0
[101] lifecycle_1.0.4 jquerylib_0.1.4
[103] cowplot_1.1.1 bitops_1.0-7
[105] irlba_2.3.5 httpuv_1.6.5
[107] rtracklayer_1.56.0 GenomicRanges_1.48.0
[109] R6_2.5.1 BiocIO_1.6.0
[111] promises_1.2.0.1 IRanges_2.30.0
[113] codetools_0.2-18 assertthat_0.2.1
[115] SummarizedExperiment_1.26.1 rprojroot_2.0.3
[117] rjson_0.2.21 withr_2.5.0
[119] GenomicAlignments_1.32.0 Rsamtools_2.12.0
[121] S4Vectors_0.34.0 GenomeInfoDbData_1.2.8
[123] parallel_4.2.0 hms_1.1.1
[125] grid_4.2.0 gggrid_0.2-0
[127] rmarkdown_2.25 MatrixGenerics_1.8.0
[129] logging_0.10-108 git2r_0.30.1
[131] mixsqp_0.3-43 Biobase_2.56.0
[133] lubridate_1.8.0 restfulr_0.0.14