Training stability weights using munro’s RNA data
XSun
2025-03-20
Last updated: 2025-03-24
Checks: 6 1
Knit directory: multigroup_ctwas_analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R
Markdown file created these results, you’ll want to first commit it to
the Git repo. If you’re still working on the analysis, you can ignore
this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20231112) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 7f6f7b1. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: cv/
Untracked files:
Untracked: analysis/data_weight_training_avg.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with
wflow_publish() to start tracking its development.
Introduction
Data source
Normalized RNA phenotype were shared by Munro et al
Covariates are downloaded from https://pantry.pejlab.org/
Genotype are from gtex v8, we imputed the missing genotype using
beagle
Workflow
Cross validation
For each RNA phenotype in each tissue, we partitioned the available samples into training (80%) and testing (20%) datasets.
- Training Phase
For each RNA molecular, we performed the following steps:
Variant Selection: We defined a ±50 kb window around the transcription start site (TSS) of each gene, following Munro et al. Variants located within this window were selected for analysis.
Preprocessing: Covariates were regressed out from both the RNA phenotype and the genotype matrix containing the selected variants. The residuals from this regression were used as inputs for subsequent analysis. (as susie tutorial described)
Fine-Mapping with SuSiE: We applied SuSiE with
L = 1andL = 5to the residualized data and identified the top variants from the credible sets.
Effect Size Estimation: For the selected variants, we re-fitted a multiple linear regression model:
model <- lm(scaled_RNA_phenotype ~ SNPs_centered + covariates_scaled) effect_sizes <- coef(model)where
scaled_RNA_phenotyperepresents the scaled RNA phenotype,SNPs_centereddenotes the centered SNP genotype matrix, andcovariates_scaledrepresents scaled covariates. The estimated coefficients from this model were used as effect sizes for the selected SNPs.
- Testing Phase
Using the trained effect sizes, we predicted RNA phenotypes in the testing dataset and evaluated model performance.
- Evaluation
We used MSE, r2 to evaluate the performance of the prediction model
r2
\[ R^2 = 1 - \frac{\sum (y_{\text{test}} - y_{\text{pred}})^2}{\sum (y_{\text{test}} - \bar{y}_{\text{test}})^2} \]
- \(y_{\text{test}}\): Observed values in the test set,
- \(y_{\text{pred}}\): Predicted values,
- \(\bar{y}_{\text{test}}\): Mean of the test set observed values.
MSE
\[ \text{MSE} = \frac{1}{n} \sum_{i=1}^n (y_i - \hat{y}_i)^2 \]
- \(y_i\): Observed value for the \(i\)-th sample,
- \(\hat{y}_i\): Predicted value for the \(i\)-th sample,
- \(n\): Total number of samples.
We performed five rounds of cross-validation and calculated the average values. Some genes had weights in certain rounds but not in others, as SuSiE did not identify credible sets. We set the threshold at 3, meaning that if a gene had an r² value from at least three rounds, we computed its mean r².”
library(RSQLite)
library(ggplot2)
library(gridExtra)
name_mapping <- read.table("/project2/xinhe/shared_data/multigroup_ctwas/weights/files_Munro/Munro_name_mapping.txt")
colnames(name_mapping) <- c("Munro_name","gtex_name")Comparing L=1 and L=5
RNA stability
qtl = "stability"
L=1
folder_pred_L1 <- paste0("/project/xinhe/xsun/weights_training/cv/multiplesets/",qtl,"/L",L,"/rsq_summary/")
folder_sample_L1<- paste0("/project/xinhe/xsun/weights_training/cv/samples/",qtl,"/")
L=5
folder_pred_L5 <- paste0("/project/xinhe/xsun/weights_training/cv/multiplesets/",qtl,"/L",L,"/rsq_summary/")
folder_sample_L5<- paste0("/project/xinhe/xsun/weights_training/cv/samples/",qtl,"/")
folder_round1 <- paste0("/project/xinhe/xsun/weights_training/cv/multiplesets/",qtl,"/L",L,"/round1/")sum <- c()
p <- list()
for (tissue in name_mapping$Munro_name) {
if(!file.exists(paste0(folder_pred_L1,tissue,"_rsq_fusion.RDS")) | !file.exists(paste0(folder_pred_L5,tissue,"_rsq_fusion.RDS"))) next
tissue_gtex <- name_mapping$gtex_name[which(name_mapping$Munro_name == tissue)]
## sample size
sample_test <- readRDS(paste0(folder_round1,tissue,"_samples_testing.RDS"))
sample_test <- length(sample_test)
sample_train <- readRDS(paste0(folder_round1,tissue,"_samples_training.RDS"))
sample_train <- length(sample_train)
sample_total <- sample_test + sample_train
## L=1
rsq_L1 <- readRDS(paste0(folder_pred_L1, tissue, "_rsq_fusion.RDS"))
n_withcs_L1 <- nrow(rsq_L1)
n_posrsq_L1 <- sum(rsq_L1$mean_rsq>0,na.rm = T)
## L=5
rsq_L5 <- readRDS(paste0(folder_pred_L5, tissue, "_rsq_fusion.RDS"))
n_withcs_L5 <- nrow(rsq_L5)
n_posrsq_L5 <- sum(rsq_L5$mean_rsq>0,na.rm = T)
n_overlap_withcs <- sum(rsq_L1$gene %in% rsq_L5$gene)
n_overlap_posrsq <- sum(rsq_L1$gene[rsq_L1$mean_rsq > 0] %in% rsq_L1$gene[rsq_L5$mean_rsq > 0])
## total rna & average qtl
weights <- readRDS(paste0(folder_round1, tissue, "_training_effectsizes.RDS"))
n_rna <- length(weights)
weights_nonnull <- Filter(Negate(is.null), weights)
avg_qtl <- mean(unlist(lapply(weights_nonnull,length)),na.rm = T)
tmp_tissue <- c(tissue_gtex,sample_total, n_rna, n_withcs_L1, n_withcs_L5,round(avg_qtl,digits = 4), n_overlap_withcs, n_posrsq_L1, n_posrsq_L5, n_overlap_posrsq)
sum <- rbind(sum, tmp_tissue)
rsq_L1_df <- data.frame(id = rsq_L1$gene, rsq_L1 = rsq_L1$mean_rsq)
rsq_L5_df <- data.frame(id = rsq_L5$gene, rsq_L5 = rsq_L5$mean_rsq)
rsq_merge <- merge(rsq_L1_df, rsq_L5_df , by = "id")
p[[tissue_gtex]] <- ggplot(rsq_merge, aes(x=rsq_L1, y=rsq_L5)) +
geom_point() +
labs(x = "rsq-L1", y="rsq-L5") +
geom_abline(slope = 1, intercept = 0, col="red") +
ggtitle(tissue_gtex) +
theme_minimal()
}
sum <- as.data.frame(sum)
rownames(sum) <- NULL
colnames(sum) <- c("tissue","sample_size_total", "n_rna_total", "n_rna_withcs_L1", "n_rna_withcs_L5", "avg_qtl_L5", "n_overlap_withcs", "n_rna_rsq+_L1", "n_rna_rsq+_L5", "n_overlap_rsq+")
sum <- sum[order(as.numeric(sum$sample_size_total),decreasing = T),]
grid.arrange(grobs = p, ncol = 4)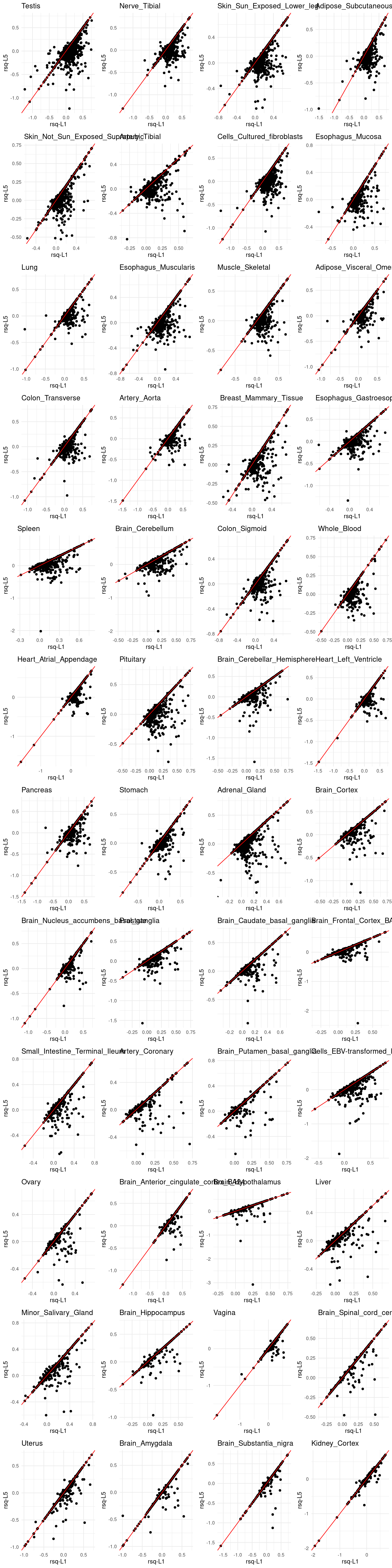
DT::datatable(sum,caption = htmltools::tags$caption( style = 'caption-side: left; text-align: left; color:black; font-size:150% ;','Comparing L=1 & L=5'),options = list(pageLength = 10) )Comparing with Munro’s prediction model
RNA stability – L=1
qtl = "stability"
L=1folder_pred <- paste0("/project/xinhe/xsun/weights_training/cv/multiplesets/",qtl,"/L",L,"/rsq_summary/")
folder_round1<- paste0("/project/xinhe/xsun/weights_training/cv/multiplesets/",qtl,"/L",L,"/round1/")
sum <- c()
for (tissue in name_mapping$Munro_name) {
if(!file.exists(paste0(folder_pred,tissue,"_rsq_fusion.RDS"))) next
tissue_gtex <- name_mapping$gtex_name[which(name_mapping$Munro_name == tissue)]
### our weights
weights <- readRDS(paste0(folder_round1, tissue, "_training_effectsizes.RDS"))
n_rna <- length(weights)
rsq <- readRDS(paste0(folder_pred, tissue, "_rsq_fusion.RDS"))
n_withcs <- nrow(rsq)
n_posrsq <- sum(rsq$mean_rsq>0,na.rm = T)
### sample size
sample_test <- readRDS(paste0(folder_round1,tissue,"_samples_testing.RDS"))
sample_test <- length(sample_test)
sample_train <- readRDS(paste0(folder_round1,tissue,"_samples_training.RDS"))
sample_train <- length(sample_train)
sample_total <- sample_test + sample_train
### Munro's weights
df_munro <- read.table(paste0("/project/xinhe/xsun/weights_training/data/weights_munro/",qtl,"/",tissue,".",qtl,".twas_weights.profile"), header = T)
n_rna_withweights_munro <- nrow(df_munro)
### overlap
n_overlap_withcs <- sum(rsq$gene %in% df_munro$id)
n_overlap_posrsq <- sum(rsq$gene[rsq$mean_rsq>0] %in% df_munro$id,na.rm=T)
tmp_tissue <- c(tissue_gtex,sample_total,n_rna,n_rna_withweights_munro, n_withcs,n_overlap_withcs,n_posrsq,n_overlap_posrsq)
sum <- rbind(sum, tmp_tissue)
}
sum <- as.data.frame(sum)
rownames(sum) <- NULL
colnames(sum) <- c("tissue","sample_size_total","n_rna_total","n_munro_weights","n_rna_withsusie_cs","overlap_cs_munro","n_rna_rsq+","overlap_rsq+_munro")
sum <- sum[order(as.numeric(sum$sample_size_total),decreasing = T),]
DT::datatable(sum,caption = htmltools::tags$caption( style = 'caption-side: left; text-align: left; color:black; font-size:150% ;','Summary for the prediction models'),options = list(pageLength = 10) )folder_munroweights<- paste0("/project/xinhe/xsun/weights_training/data/weights_munro/",qtl,"/")
p <- list()
for (tissue in name_mapping$Munro_name) {
if(!file.exists(paste0(folder_pred,tissue,"_rsq_fusion.RDS"))) next
tissue_gtex <- name_mapping$gtex_name[which(name_mapping$Munro_name == tissue)]
### our weights
rsq <- readRDS(paste0(folder_pred, tissue, "_rsq_fusion.RDS"))
rsq <- data.frame(id = rsq$gene, rsq = rsq$mean_rsq)
### Munro's weights
summary_munro <- read.table(paste0(folder_munroweights,tissue,".",qtl,".twas_weights.profile"),header = T)
merged_df <- merge(rsq, summary_munro, by= "id")
p[[tissue_gtex]] <- ggplot(merged_df, aes(x=rsq, y=lasso.r2)) +
geom_point() +
labs(x = "rsq-topsnpfromsusie", y="rsq-lasso-munro") +
geom_abline(slope = 1, intercept = 0, col="red") +
ggtitle(tissue_gtex) +
theme_minimal()
}
grid.arrange(grobs = p, ncol = 4)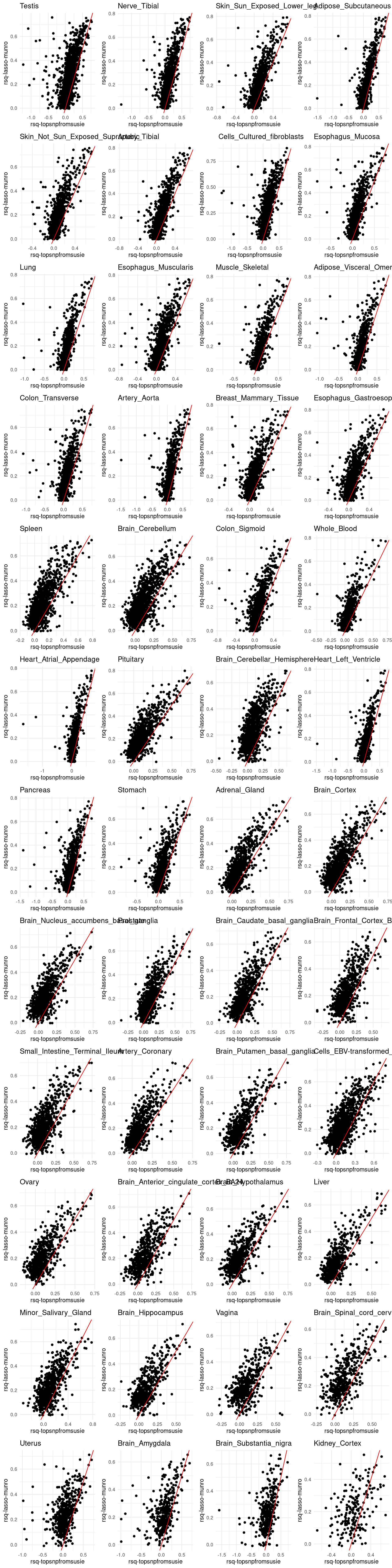
RNA stability – L=5
qtl = "stability"
L=5folder_pred <- paste0("/project/xinhe/xsun/weights_training/cv/multiplesets/",qtl,"/L",L,"/rsq_summary/")
folder_round1<- paste0("/project/xinhe/xsun/weights_training/cv/multiplesets/",qtl,"/L",L,"/round1/")
sum <- c()
for (tissue in name_mapping$Munro_name) {
if(!file.exists(paste0(folder_pred,tissue,"_rsq_fusion.RDS"))) next
tissue_gtex <- name_mapping$gtex_name[which(name_mapping$Munro_name == tissue)]
### our weights
weights <- readRDS(paste0(folder_round1, tissue, "_training_effectsizes.RDS"))
n_rna <- length(weights)
rsq <- readRDS(paste0(folder_pred, tissue, "_rsq_fusion.RDS"))
n_withcs <- nrow(rsq)
n_posrsq <- sum(rsq$mean_rsq>0,na.rm = T)
### sample size
sample_test <- readRDS(paste0(folder_round1,tissue,"_samples_testing.RDS"))
sample_test <- length(sample_test)
sample_train <- readRDS(paste0(folder_round1,tissue,"_samples_training.RDS"))
sample_train <- length(sample_train)
sample_total <- sample_test + sample_train
### Munro's weights
df_munro <- read.table(paste0("/project/xinhe/xsun/weights_training/data/weights_munro/",qtl,"/",tissue,".",qtl,".twas_weights.profile"), header = T)
n_rna_withweights_munro <- nrow(df_munro)
### overlap
n_overlap_withcs <- sum(rsq$gene %in% df_munro$id)
n_overlap_posrsq <- sum(rsq$gene[rsq$mean_rsq>0] %in% df_munro$id,na.rm=T)
tmp_tissue <- c(tissue_gtex,sample_total,n_rna,n_rna_withweights_munro, n_withcs,n_overlap_withcs,n_posrsq,n_overlap_posrsq)
sum <- rbind(sum, tmp_tissue)
}
sum <- as.data.frame(sum)
rownames(sum) <- NULL
colnames(sum) <- c("tissue","sample_size_total","n_rna_total","n_munro_weights","n_rna_withsusie_cs","overlap_cs_munro","n_rna_rsq+","overlap_rsq+_munro")
sum <- sum[order(as.numeric(sum$sample_size_total),decreasing = T),]
DT::datatable(sum,caption = htmltools::tags$caption( style = 'caption-side: left; text-align: left; color:black; font-size:150% ;','Summary for the prediction models'),options = list(pageLength = 10) )folder_munroweights<- paste0("/project/xinhe/xsun/weights_training/data/weights_munro/",qtl,"/")
p <- list()
for (tissue in name_mapping$Munro_name) {
if(!file.exists(paste0(folder_pred,tissue,"_rsq_fusion.RDS"))) next
tissue_gtex <- name_mapping$gtex_name[which(name_mapping$Munro_name == tissue)]
### our weights
rsq <- readRDS(paste0(folder_pred, tissue, "_rsq_fusion.RDS"))
rsq <- data.frame(id = rsq$gene, rsq = rsq$mean_rsq)
### Munro's weights
summary_munro <- read.table(paste0(folder_munroweights,tissue,".",qtl,".twas_weights.profile"),header = T)
merged_df <- merge(rsq, summary_munro, by= "id")
p[[tissue_gtex]] <- ggplot(merged_df, aes(x=rsq, y=lasso.r2)) +
geom_point() +
labs(x = "rsq-topsnpfromsusie", y="rsq-lasso-munro") +
geom_abline(slope = 1, intercept = 0, col="red") +
ggtitle(tissue_gtex) +
theme_minimal()
}
grid.arrange(grobs = p, ncol = 4)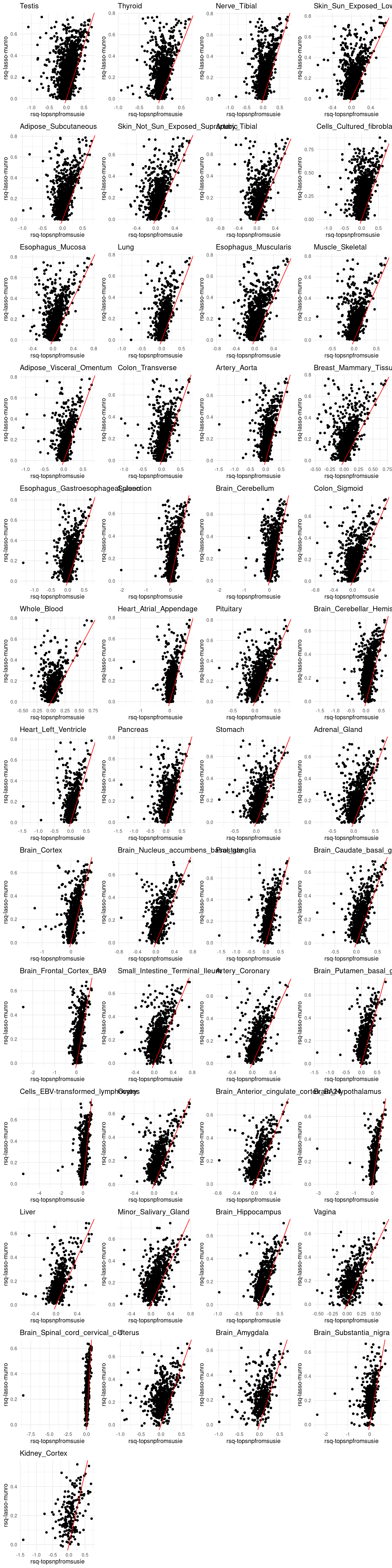
sessionInfo()R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] gridExtra_2.3 ggplot2_3.5.1 RSQLite_2.3.1
loaded via a namespace (and not attached):
[1] tidyselect_1.2.0 xfun_0.41 bslib_0.3.1 colorspace_2.0-3
[5] vctrs_0.6.5 generics_0.1.2 htmltools_0.5.2 yaml_2.3.5
[9] utf8_1.2.2 blob_1.2.3 rlang_1.1.2 jquerylib_0.1.4
[13] later_1.3.0 pillar_1.9.0 glue_1.6.2 withr_2.5.0
[17] DBI_1.2.2 bit64_4.0.5 lifecycle_1.0.4 stringr_1.5.1
[21] munsell_0.5.0 gtable_0.3.0 workflowr_1.7.0 htmlwidgets_1.5.4
[25] evaluate_0.15 memoise_2.0.1 labeling_0.4.2 knitr_1.39
[29] fastmap_1.1.0 httpuv_1.6.5 crosstalk_1.2.0 fansi_1.0.3
[33] highr_0.9 Rcpp_1.0.12 promises_1.2.0.1 scales_1.3.0
[37] DT_0.22 cachem_1.0.6 jsonlite_1.8.0 farver_2.1.0
[41] fs_1.5.2 bit_4.0.4 digest_0.6.29 stringi_1.7.6
[45] dplyr_1.1.4 rprojroot_2.0.3 grid_4.2.0 cli_3.6.1
[49] tools_4.2.0 magrittr_2.0.3 sass_0.4.1 tibble_3.2.1
[53] pkgconfig_2.0.3 rmarkdown_2.25 rstudioapi_0.13 R6_2.5.1
[57] git2r_0.30.1 compiler_4.2.0