Simulation Results using three Tissues (correlated)
shengqian
2023-10-13
Last updated: 2023-10-13
Checks: 6 1
Knit directory: cTWAS_analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20211220) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 68eacf2. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .ipynb_checkpoints/
Untracked files:
Untracked: LDL_LDLR_locus1.pdf
Untracked: LDL_TEME199_genetrack.pdf
Untracked: LDL_TEME199_locus.pdf
Untracked: Proposal plots.R
Untracked: RGS14.pdf
Untracked: RNF186.pdf
Untracked: Rplots.pdf
Untracked: SCZ_annotation.xlsx
Untracked: SLC8B1.pdf
Untracked: analysis/.ipynb_checkpoints/
Untracked: analysis/OxHB.Rmd
Untracked: analysis/simulation_3tissues_correlated.Rmd
Untracked: analysis/simulation_3tissues_uncorrelated.Rmd
Untracked: cache/
Untracked: code/.ipynb_checkpoints/
Untracked: code/OxHb_out/OxHb_Kidney_Cortex.err
Untracked: code/OxHb_out/OxHb_Kidney_Cortex.out
Untracked: code/OxHb_out/OxHb_Liver.err
Untracked: code/OxHb_out/OxHb_Liver.out
Untracked: code/OxHb_out/OxHb_Pancreas.err
Untracked: code/OxHb_out/OxHb_Pancreas.out
Untracked: code/OxHb_out/OxHb_Whole_Blood.err
Untracked: code/OxHb_out/OxHb_Whole_Blood.out
Untracked: data/.ipynb_checkpoints/
Untracked: data/FUMA_output/
Untracked: data/GO_Terms/
Untracked: data/GTEx_Analysis_v8_eQTL.tar
Untracked: data/G_list.RData
Untracked: data/IBD_ME/
Untracked: data/LB/
Untracked: data/LDL/
Untracked: data/LDL_E_S/
Untracked: data/LDL_E_S_M/
Untracked: data/LDL_M/
Untracked: data/LDL_S/
Untracked: data/OxHb/
Untracked: data/PGC3_SCZ_wave3_public.v2.tsv
Untracked: data/Predictive_Models/
Untracked: data/Supplementary Table 15 - MAGMA.xlsx
Untracked: data/Supplementary Table 20 - Prioritised Genes.xlsx
Untracked: data/UKBB/
Untracked: data/UKBB_SNPs_Info.text
Untracked: data/WhiteBlood_E/
Untracked: data/WhiteBlood_E_M/
Untracked: data/WhiteBlood_E_S_M/
Untracked: data/WhiteBlood_E_S_M_PC/
Untracked: data/WhiteBlood_M/
Untracked: data/WhiteBlood_M_compare/
Untracked: data/WhiteBlood_M_enet/
Untracked: data/cpg_annot.RData
Untracked: data/eqtl/
Untracked: data/gencode.v26.GRCh38.genes.gtf
Untracked: data/gene_OMIM.txt
Untracked: data/gene_pip_0.8.txt
Untracked: data/gwas_sumstats/
Untracked: data/magma.genes.out
Untracked: data/mashr_Heart_Atrial_Appendage.db
Untracked: data/mashr_sqtl/
Untracked: data/mqtl/
Untracked: data/notes.txt
Untracked: data/scz_2018.RDS
Untracked: data/summary_known_genes_annotations.xlsx
Untracked: data/test/
Untracked: hist.pdf
Untracked: ld_pip.pdf
Untracked: submit.sh
Untracked: temp_LDR/
Untracked: test-B1.snpgwas.txt
Unstaged changes:
Deleted: analysis/Atrial_Fibrillation_Heart_Atrial_Appendage.Rmd
Deleted: analysis/Atrial_Fibrillation_Heart_Left_Ventricle.Rmd
Deleted: analysis/Autism_Brain_Amygdala.Rmd
Deleted: analysis/Autism_Brain_Anterior_cingulate_cortex_BA24.Rmd
Deleted: analysis/Autism_Brain_Caudate_basal_ganglia.Rmd
Deleted: analysis/Autism_Brain_Cerebellar_Hemisphere.Rmd
Deleted: analysis/Autism_Brain_Cerebellum.Rmd
Deleted: analysis/Autism_Brain_Cortex.Rmd
Deleted: analysis/Autism_Brain_Frontal_Cortex_BA9.Rmd
Deleted: analysis/Autism_Brain_Hippocampus.Rmd
Deleted: analysis/Autism_Brain_Hypothalamus.Rmd
Deleted: analysis/Autism_Brain_Nucleus_accumbens_basal_ganglia.Rmd
Deleted: analysis/Autism_Brain_Putamen_basal_ganglia.Rmd
Deleted: analysis/Autism_Brain_Spinal_cord_cervical_c-1.Rmd
Deleted: analysis/Autism_Brain_Substantia_nigra.Rmd
Deleted: analysis/BMI_Brain_Amygdala.Rmd
Deleted: analysis/BMI_Brain_Amygdala_S.Rmd
Deleted: analysis/BMI_Brain_Anterior_cingulate_cortex_BA24.Rmd
Deleted: analysis/BMI_Brain_Anterior_cingulate_cortex_BA24_S.Rmd
Deleted: analysis/BMI_Brain_Caudate_basal_ganglia.Rmd
Deleted: analysis/BMI_Brain_Caudate_basal_ganglia_S.Rmd
Deleted: analysis/BMI_Brain_Cerebellar_Hemisphere.Rmd
Deleted: analysis/BMI_Brain_Cerebellar_Hemisphere_S.Rmd
Deleted: analysis/BMI_Brain_Cerebellum.Rmd
Deleted: analysis/BMI_Brain_Cerebellum_S.Rmd
Deleted: analysis/BMI_Brain_Cortex.Rmd
Deleted: analysis/BMI_Brain_Cortex_S.Rmd
Deleted: analysis/BMI_Brain_Frontal_Cortex_BA9.Rmd
Deleted: analysis/BMI_Brain_Frontal_Cortex_BA9_S.Rmd
Deleted: analysis/BMI_Brain_Hippocampus.Rmd
Deleted: analysis/BMI_Brain_Hippocampus_S.Rmd
Deleted: analysis/BMI_Brain_Hypothalamus.Rmd
Deleted: analysis/BMI_Brain_Hypothalamus_S.Rmd
Deleted: analysis/BMI_Brain_Nucleus_accumbens_basal_ganglia.Rmd
Deleted: analysis/BMI_Brain_Nucleus_accumbens_basal_ganglia_S.Rmd
Deleted: analysis/BMI_Brain_Putamen_basal_ganglia.Rmd
Deleted: analysis/BMI_Brain_Putamen_basal_ganglia_S.Rmd
Deleted: analysis/BMI_Brain_Spinal_cord_cervical_c-1.Rmd
Deleted: analysis/BMI_Brain_Spinal_cord_cervical_c-1_S.Rmd
Deleted: analysis/BMI_Brain_Substantia_nigra.Rmd
Deleted: analysis/BMI_Brain_Substantia_nigra_S.Rmd
Deleted: analysis/BMI_S_results.Rmd
Deleted: analysis/Glucose_Adipose_Subcutaneous.Rmd
Deleted: analysis/Glucose_Adipose_Visceral_Omentum.Rmd
Modified: analysis/index.Rmd
Deleted: code/OxHb_out/OxHb_Artery_Aorta.err
Deleted: code/OxHb_out/OxHb_Artery_Aorta.out
Modified: code/OxHb_out/OxHb_Artery_Coronary.err
Modified: code/OxHb_out/OxHb_Artery_Coronary.out
Modified: code/OxHb_out/OxHb_Artery_Tibial.out
Modified: code/OxHb_out/OxHb_Heart_Atrial_Appendage.err
Modified: code/OxHb_out/OxHb_Heart_Atrial_Appendage.out
Modified: code/OxHb_out/OxHb_Heart_Left_Ventricle.out
Modified: code/OxHb_out/OxHb_Lung.err
Modified: code/OxHb_out/OxHb_Lung.out
Deleted: code/WhiteBlood_M_ener_out/WhiteBlood_WholeBlood.out
Deleted: code/White_Blood_M_out/White_Blood_BreastMammary.err
Deleted: code/White_Blood_M_out/White_Blood_BreastMammary.out
Deleted: code/White_Blood_M_out/White_Blood_ColonTransverse.err
Deleted: code/White_Blood_M_out/White_Blood_ColonTransverse.out
Deleted: code/White_Blood_M_out/White_Blood_KidneyCortex.err
Deleted: code/White_Blood_M_out/White_Blood_KidneyCortex.out
Deleted: code/White_Blood_M_out/White_Blood_Lung.err
Deleted: code/White_Blood_M_out/White_Blood_Lung.out
Deleted: code/White_Blood_M_out/White_Blood_MuscleSkeletal.err
Deleted: code/White_Blood_M_out/White_Blood_MuscleSkeletal.out
Deleted: code/White_Blood_M_out/White_Blood_Ovary.err
Deleted: code/White_Blood_M_out/White_Blood_Ovary.out
Deleted: code/White_Blood_M_out/White_Blood_Prostate.err
Deleted: code/White_Blood_M_out/White_Blood_Prostate.out
Deleted: code/White_Blood_M_out/White_Blood_Testis.err
Deleted: code/White_Blood_M_out/White_Blood_Testis.out
Deleted: code/White_Blood_M_out/White_Blood_WholeBlood.err
Deleted: code/White_Blood_M_out/White_Blood_WholeBlood.out
Deleted: code/run_IBD_ctwas_rss_LDR_ME.R
Modified: code/run_OxHb_analysis.sh
Modified: code/run_OxHb_ctwas_rss_LDR.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Simulation 1: Two causal tissues with equal high PVE
Separate effect size parameters
For the cTWAS analysis, each tissue had its own prior inclusion parameter end effect size parameter.
results_dir <- "/project2/xinhe/shengqian/cTWAS/cTWAS_simulation/simulation_correlated/"
runtag = "ukb-s80.45-3_corr"
configtag <- 2
simutags <- paste(1, 1:3, sep = "-")
thin <- 0.1
sample_size <- 45000
PIP_threshold <- 0.8results_df <- data.frame(simutag=as.character(),
n_causal=as.integer(),
n_causal_combined=as.integer(),
n_detected_pip=as.integer(),
n_detected_pip_in_causal=as.integer(),
n_detected_comb_pip=as.integer(),
n_detected_comb_pip_in_causal=as.integer(),
pve_snp=as.numeric(),
pve_weight1=as.numeric(),
pve_weight2=as.numeric(),
pve_weight3=as.numeric(),
prior_weight1=as.numeric(),
prior_weight2=as.numeric(),
prior_weight3=as.numeric(),
prior_var_snp=as.numeric(),
prior_var_weight1=as.numeric(),
prior_var_weight2=as.numeric(),
prior_var_weight3=as.numeric())
for (i in 1:length(simutags)){
simutag <- simutags[i]
#load genes with true simulated effect
load(paste0(results_dir, runtag, "_simu", simutag, "-pheno.Rd"))
true_genes <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$id.cgene}))
true_genes_combined <- unique(sapply(true_genes, function(x){unlist(strsplit(x, "[|]"))[1]}))
#load cTWAS results
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of causal genes
n_causal <- length(true_genes)
n_causal_combined <- length(true_genes_combined)
#number of gene+tissue combinations with cTWAS PIP > threshold
n_ctwas_genes <- sum(ctwas_gene_res$susie_pip > PIP_threshold)
#number of cTWAS genes that are causal
n_causal_detected <- sum(ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold] %in% true_genes)
#collapse gene+tissues to genes and compute combined PIP
ctwas_gene_res$gene <- sapply(ctwas_gene_res$id, function(x){unlist(strsplit(x,"[|]"))[1]})
ctwas_gene_res_combined <- aggregate(ctwas_gene_res$susie_pip, by=list(ctwas_gene_res$gene), FUN=sum)
colnames(ctwas_gene_res_combined) <- c("gene", "pip_combined")
#number of genes with combined PIP > threshold
n_ctwas_genes_combined <- sum(ctwas_gene_res_combined$pip_combined > PIP_threshold)
#number of cTWAS genes using combined PIP that are causal
n_causal_detected_combined <- sum(ctwas_gene_res_combined$gene[ctwas_gene_res_combined$pip_combined > PIP_threshold] %in% true_genes_combined)
#collect number of SNPs analyzed by cTWAS
ctwas_res_s1 <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.s1.susieIrss.txt"))
n_snps <- sum(ctwas_res_s1$type=="SNP")/thin
rm(ctwas_res_s1)
#load estimated parameters
load(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.s2.susieIrssres.Rd"))
#estimated group prior (all iterations)
estimated_group_prior_all <- group_prior_rec
estimated_group_prior_all["SNP",] <- estimated_group_prior_all["SNP",]*thin #adjust parameter to account for thin argument
#estimated group prior variance (all iterations)
estimated_group_prior_var_all <- group_prior_var_rec
#set group size
group_size <- c(table(ctwas_gene_res$type), structure(n_snps, names="SNP"))
group_size <- group_size[rownames(estimated_group_prior_all)]
#estimated group PVE (all iterations)
estimated_group_pve_all <- estimated_group_prior_var_all*estimated_group_prior_all*group_size/sample_size #check PVE calculation
results_current <- data.frame(simutag=as.character(simutag),
n_causal=as.integer(n_causal),
n_causal_combined=as.integer(n_causal_combined),
n_detected_pip=as.integer(n_ctwas_genes),
n_detected_pip_in_causal=as.integer(n_causal_detected),
n_detected_comb_pip=as.integer(n_ctwas_genes_combined),
n_detected_comb_pip_in_causal=as.integer(n_causal_detected_combined),
pve_snp=as.numeric(rev(estimated_group_pve_all["SNP",])[1]),
pve_weight1=as.numeric(rev(estimated_group_pve_all["Brain_Cerebellum_harmonized",])[1]),
pve_weight2=as.numeric(rev(estimated_group_pve_all["Brain_Hippocampus_harmonized",])[1]),
pve_weight3=as.numeric(rev(estimated_group_pve_all["Brain_Caudate_basal_ganglia_harmonized",])[1]),
prior_snp=as.numeric(rev(estimated_group_prior_var_all["SNP",])[1]),
prior_weight1=as.numeric(rev(estimated_group_prior_all["Brain_Cerebellum_harmonized",])[1]),
prior_weight2=as.numeric(rev(estimated_group_prior_all["Brain_Hippocampus_harmonized",])[1]),
prior_weight3=as.numeric(rev(estimated_group_prior_all["Brain_Caudate_basal_ganglia_harmonized",])[1]),
prior_var_snp=as.numeric(rev(estimated_group_prior_var_all["SNP",])[1]),
prior_var_weight1=as.numeric(rev(estimated_group_prior_var_all["Brain_Cerebellum_harmonized",])[1]),
prior_var_weight2=as.numeric(rev(estimated_group_prior_var_all["Brain_Hippocampus_harmonized",])[1]),
prior_var_weight3=as.numeric(rev(estimated_group_prior_var_all["Brain_Caudate_basal_ganglia_harmonized",])[1]))
results_df <- rbind(results_df, results_current)
}
#results using PIP threshold (gene+tissue)
results_df[,c("simutag", "n_causal", "n_detected_pip", "n_detected_pip_in_causal")] simutag n_causal n_detected_pip n_detected_pip_in_causal
1 1-1 217 58 17
2 1-2 238 114 41
3 1-3 208 83 28#mean percent causal using PIP > 0.8
sum(results_df$n_detected_pip_in_causal)/sum(results_df$n_detected_pip)[1] 0.3372549#results using combined PIP threshold
results_df[,c("simutag", "n_causal_combined", "n_detected_comb_pip", "n_detected_comb_pip_in_causal")] simutag n_causal_combined n_detected_comb_pip n_detected_comb_pip_in_causal
1 1-1 215 64 31
2 1-2 236 123 53
3 1-3 206 86 38#mean percent causal using combined PIP > 0.8
sum(results_df$n_detected_comb_pip_in_causal)/sum(results_df$n_detected_comb_pip)[1] 0.4468864#prior inclusion and mean prior inclusion
results_df[,c(which(colnames(results_df)=="simutag"), setdiff(grep("prior", names(results_df)), grep("prior_var", names(results_df))))] simutag prior_snp prior_weight1 prior_weight2 prior_weight3
1 1-1 9.260675 0.004795843 0.008183196 0.015153959
2 1-2 7.972147 0.008051228 0.006912857 0.004554866
3 1-3 9.160136 0.014422878 0.005114593 0.007250383colMeans(results_df[,setdiff(grep("prior", names(results_df)), grep("prior_var", names(results_df)))]) prior_snp prior_weight1 prior_weight2 prior_weight3
8.797652886 0.009089983 0.006736882 0.008986402 #prior variance and mean prior variance
results_df[,c(which(colnames(results_df)=="simutag"), grep("prior_var", names(results_df)))] simutag prior_var_snp prior_var_weight1 prior_var_weight2 prior_var_weight3
1 1-1 9.260675 40.75581 22.65020 7.843306
2 1-2 7.972147 20.75719 36.76933 33.636322
3 1-3 9.160136 11.06991 15.12579 27.476631colMeans(results_df[,grep("prior_var", names(results_df))]) prior_var_snp prior_var_weight1 prior_var_weight2 prior_var_weight3
8.797653 24.194304 24.848437 22.985420 #PVE and mean PVE
results_df[,c(which(colnames(results_df)=="simutag"), grep("pve", names(results_df)))] simutag pve_snp pve_weight1 pve_weight2 pve_weight3
1 1-1 0.2993075 0.03809702 0.03274535 0.02266473
2 1-2 0.2733422 0.03257372 0.04490532 0.02921524
3 1-3 0.2932229 0.03111951 0.01366733 0.03798830colMeans(results_df[,grep("pve", names(results_df))]) pve_snp pve_weight1 pve_weight2 pve_weight3
0.28862420 0.03393008 0.03043933 0.02995609 #store results for figure
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS G - Ind Var",
count=results_df$n_detected_comb_pip-results_df$n_detected_comb_pip_in_causal,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS G - Ind Var",
count=results_df$n_detected_comb_pip_in_causal,
ifcausal=T))Individual tissue analyses
For the cTWAS analysis, each tissue was analyzed individually and the results were combined.
results_dir <- "/project2/xinhe/shengqian/cTWAS/cTWAS_simulation/simulation_correlated/"
runtag = "ukb-s80.45-3_corr"
configtag <- 2
simutags <- paste(1, 1:3, sep = "-")
thin <- 0.1
sample_size <- 45000
PIP_threshold <- 0.8
results_df <- data.frame(simutag=as.character(),
n_causal_combined=as.integer(),
n_detected_weight1=as.integer(),
n_detected_in_causal_weight1=as.integer(),
n_detected_weight2=as.integer(),
n_detected_in_causal_weight2=as.integer(),
n_detected_weight3=as.integer(),
n_detected_in_causal_weight3=as.integer(),
n_detected_combined=as.integer(),
n_detected_in_causal_combined=as.integer())
for (i in 1:length(simutags)){
simutag <- simutags[i]
#load genes with true simulated effect
load(paste0(results_dir, runtag, "_simu", simutag, "-pheno.Rd"))
true_genes <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$id.cgene}))
true_genes_combined <- unique(sapply(true_genes, function(x){unlist(strsplit(x, "[|]"))[1]}))
#number of causal genes
n_causal_combined <- length(true_genes_combined)
#load cTWAS results for weight1
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight1.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight1
ctwas_genes_weight1 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight1 <- length(ctwas_genes_weight1)
n_causal_detected_weight1 <- sum(ctwas_genes_weight1 %in% true_genes_combined)
#load cTWAS results for weight2
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight2.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight1
ctwas_genes_weight2 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight2 <- length(ctwas_genes_weight2)
n_causal_detected_weight2 <- sum(ctwas_genes_weight2 %in% true_genes_combined)
#load cTWAS results for weight3
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight3.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight3
ctwas_genes_weight3 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight3 <- length(ctwas_genes_weight3)
n_causal_detected_weight3 <- sum(ctwas_genes_weight3 %in% true_genes_combined)
#combined analysis
ctwas_genes_combined <- unique(c(ctwas_genes_weight1, ctwas_genes_weight2, ctwas_genes_weight3))
n_ctwas_genes_combined <- length(ctwas_genes_combined)
n_causal_detected_combined <- sum(ctwas_genes_combined %in% true_genes_combined)
results_current <- data.frame(simutag=as.character(simutag),
n_causal_combined=as.integer(n_causal_combined),
n_detected_weight1=as.integer(n_ctwas_genes_weight1),
n_detected_in_causal_weight1=as.integer(n_causal_detected_weight1),
n_detected_weight2=as.integer(n_ctwas_genes_weight2),
n_detected_in_causal_weight2=as.integer(n_causal_detected_weight2),
n_detected_weight3=as.integer(n_ctwas_genes_weight3),
n_detected_in_causal_weight3=as.integer(n_causal_detected_weight3),
n_detected_combined=as.integer(n_ctwas_genes_combined),
n_detected_in_causal_combined=as.integer(n_causal_detected_combined))
results_df <- rbind(results_df, results_current)
}
#results using weight1
results_df[,c("simutag", colnames(results_df)[grep("weight1", colnames(results_df))])] simutag n_detected_weight1 n_detected_in_causal_weight1
1 1-1 37 18
2 1-2 68 32
3 1-3 24 11#mean percent causal using PIP > 0.8 for weight1
sum(results_df$n_detected_in_causal_weight1)/sum(results_df$n_detected_weight1)[1] 0.4728682#results using weight2
results_df[,c("simutag", colnames(results_df)[grep("weight2", colnames(results_df))])] simutag n_detected_weight2 n_detected_in_causal_weight2
1 1-1 42 21
2 1-2 49 19
3 1-3 42 19#mean percent causal using PIP > 0.8 for weight1
sum(results_df$n_detected_in_causal_weight2)/sum(results_df$n_detected_weight2)[1] 0.443609#results using weight3
results_df[,c("simutag", colnames(results_df)[grep("weight3", colnames(results_df))])] simutag n_detected_weight3 n_detected_in_causal_weight3
1 1-1 39 18
2 1-2 56 23
3 1-3 0 0#mean percent causal using PIP > 0.8 for weight3
sum(results_df$n_detected_in_causal_weight3)/sum(results_df$n_detected_weight3)[1] 0.4315789#results using combined analysis
results_df[,c("simutag", colnames(results_df)[grep("combined", colnames(results_df))])] simutag n_causal_combined n_detected_combined n_detected_in_causal_combined
1 1-1 215 80 35
2 1-2 236 124 49
3 1-3 206 58 26#mean percent causal using PIP > 0.8 for weight3
sum(results_df$n_detected_in_causal_combined)/sum(results_df$n_detected_combined)[1] 0.4198473#store results for figure
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight1",
count=results_df$n_detected_weight1-results_df$n_detected_in_causal_weight1,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight1",
count=results_df$n_detected_in_causal_weight1,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight2",
count=results_df$n_detected_weight2-results_df$n_detected_in_causal_weight2,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight2",
count=results_df$n_detected_in_causal_weight2,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight3",
count=results_df$n_detected_weight3-results_df$n_detected_in_causal_weight3,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight3",
count=results_df$n_detected_in_causal_weight3,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Union",
count=results_df$n_detected_combined-results_df$n_detected_in_causal_combined,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Union",
count=results_df$n_detected_in_causal_combined,
ifcausal=T))Figure
For the cTWAS analysis, each tissue was analyzed individually and the results were combined.
plot_df$method <- factor(plot_df$method, levels=c("TWAS", "cTWAS G+T", "cTWAS G", "cTWAS G - Ind Var", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3"))
library(ggpubr)Loading required package: ggplot2colset = c("#ebebeb", "#fb8072")
ggbarplot(plot_df,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "none",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1))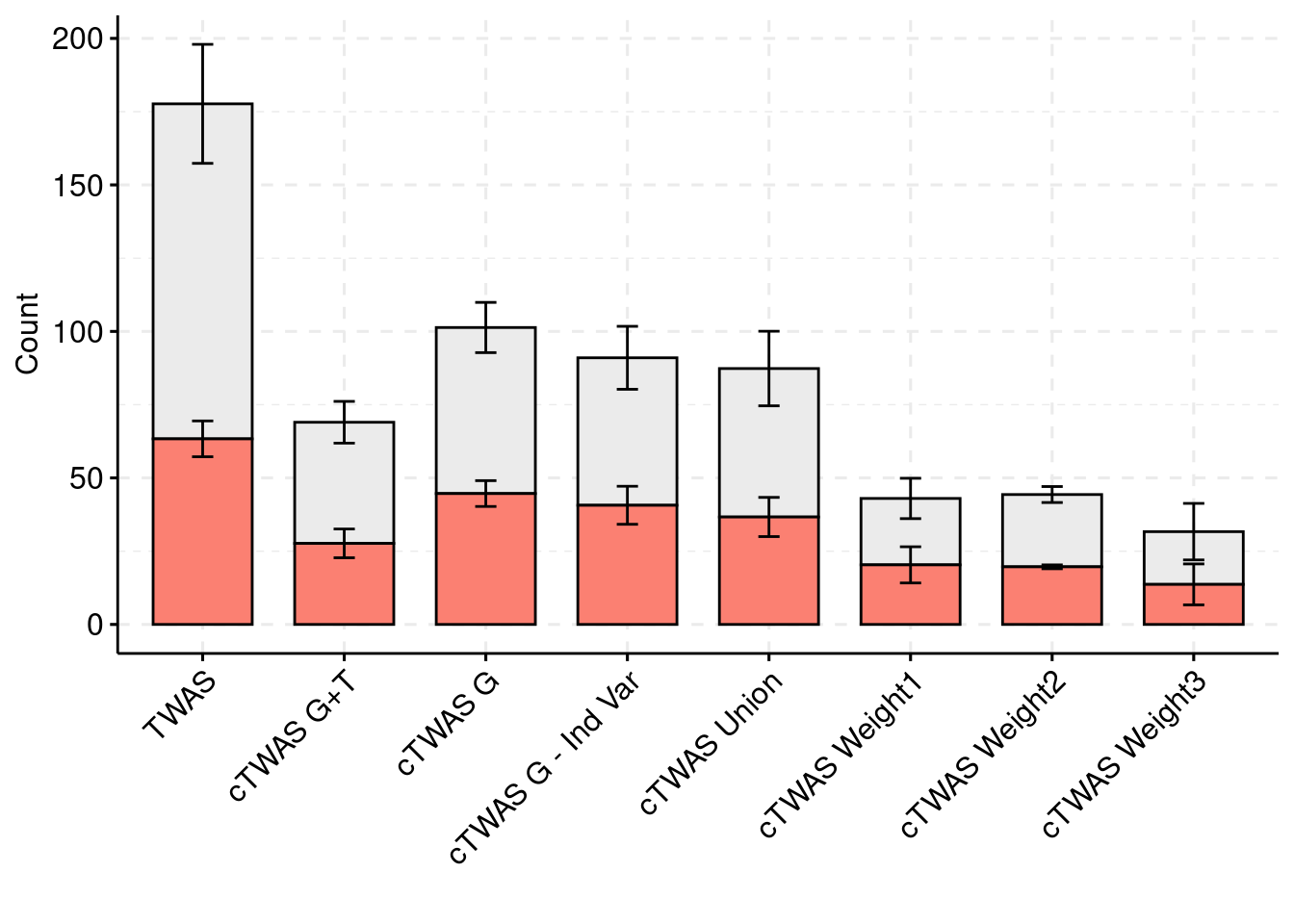
plot_df_2 <- plot_df
plot_df_2$method %in% c("TWAS", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3") [1] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE
[13] TRUE TRUE TRUE TRUE TRUE TRUE FALSE FALSE FALSE FALSE FALSE FALSE
[25] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
[37] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUEplot_df_2$ifcausal <- as.character(plot_df_2$ifcausal+1)
plot_df_2$count[plot_df_2$method=="cTWAS G" & plot_df_2$ifcausal=="2"] <- plot_df_2$count[plot_df_2$method=="cTWAS G" & plot_df_2$ifcausal=="2"] - plot_df_2$count[plot_df_2$method=="cTWAS G+T" & plot_df_2$ifcausal=="2"]
plot_df_2 <- rbind(plot_df_2, data.frame(simutag=simutags,
method="cTWAS G",
count=plot_df_2$count[plot_df_2$method=="cTWAS G+T" & plot_df_2$ifcausal=="2"],
ifcausal="3"))
plot_df_3 <- plot_df_2
plot_df_2 <- plot_df_2[plot_df_2$method %in% c("TWAS", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3"),]
plot_df_2$method <- droplevels(plot_df_2$method)
levels(plot_df_2$method) <- c("TWAS", "cTWAS Union", "cTWAS Primary", "cTWAS Secondary", "cTWAS Null")
plot_df_2$ifcausal[plot_df_2$ifcausal=="1"] <- "FP"
plot_df_2$ifcausal[plot_df_2$ifcausal=="2"] <- "TP"
colset = c("#ebebeb", "#fb8072", "#8dd3c7")
ggbarplot(plot_df_2,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "right",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1)) + guides(fill=guide_legend(title=""))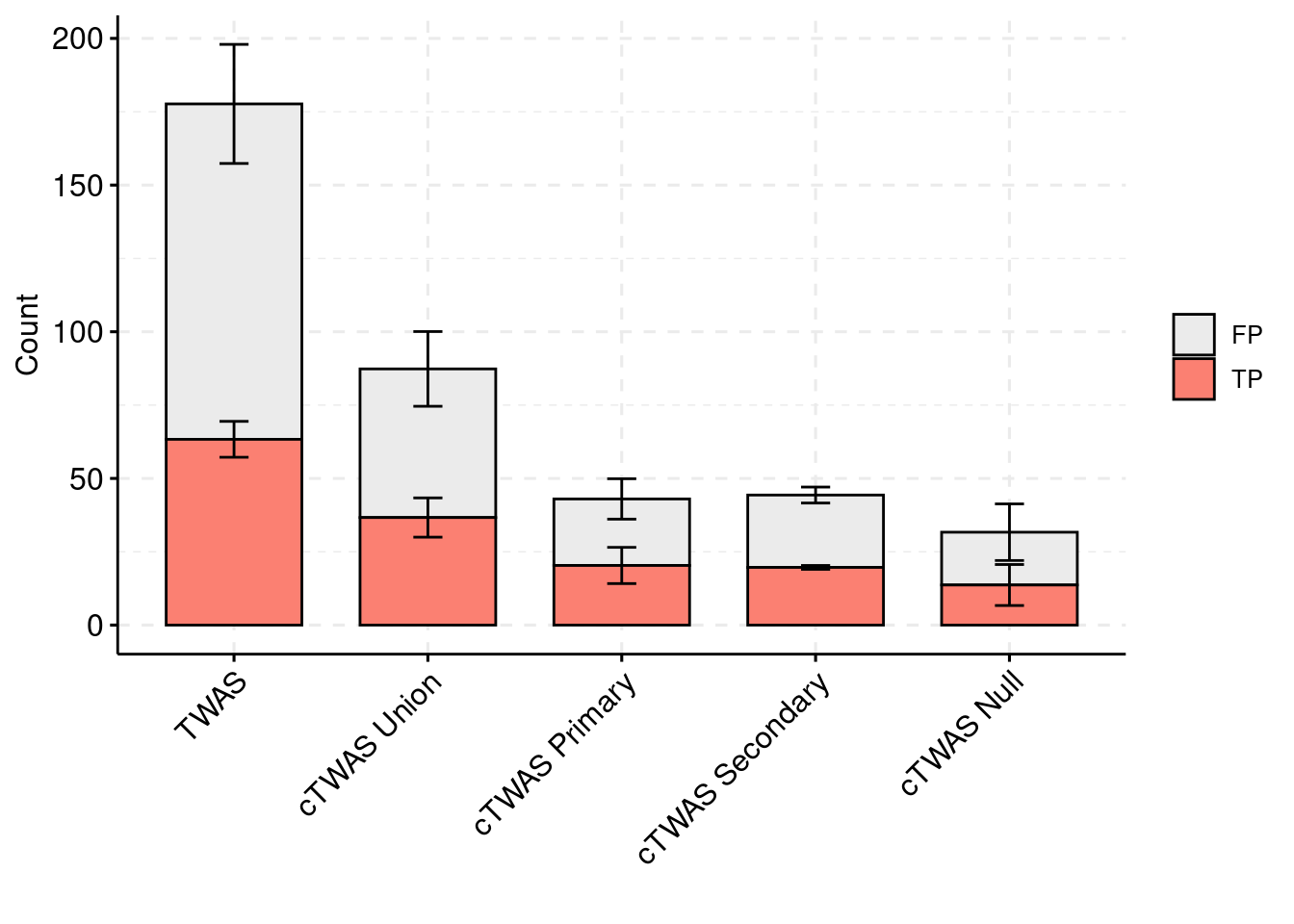
plot_df_3 <- plot_df_3[plot_df_3$method %in% c("cTWAS G", "cTWAS Union"),]
plot_df_3$method <- droplevels(plot_df_3$method)
levels(plot_df_3$method) <- c("cTWAS Joint", "cTWAS Union")
plot_df_3$method <- relevel(plot_df_3$method, "cTWAS Union")
plot_df_3$ifcausal[plot_df_3$ifcausal=="1"] <- "FP"
plot_df_3$ifcausal[plot_df_3$ifcausal=="2"] <- "TP"
plot_df_3$ifcausal[plot_df_3$ifcausal=="3"] <- "TP and Tissue Identified\nby Joint Analysis"
ggbarplot(plot_df_3,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "right",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1)) + guides(fill=guide_legend(title=""))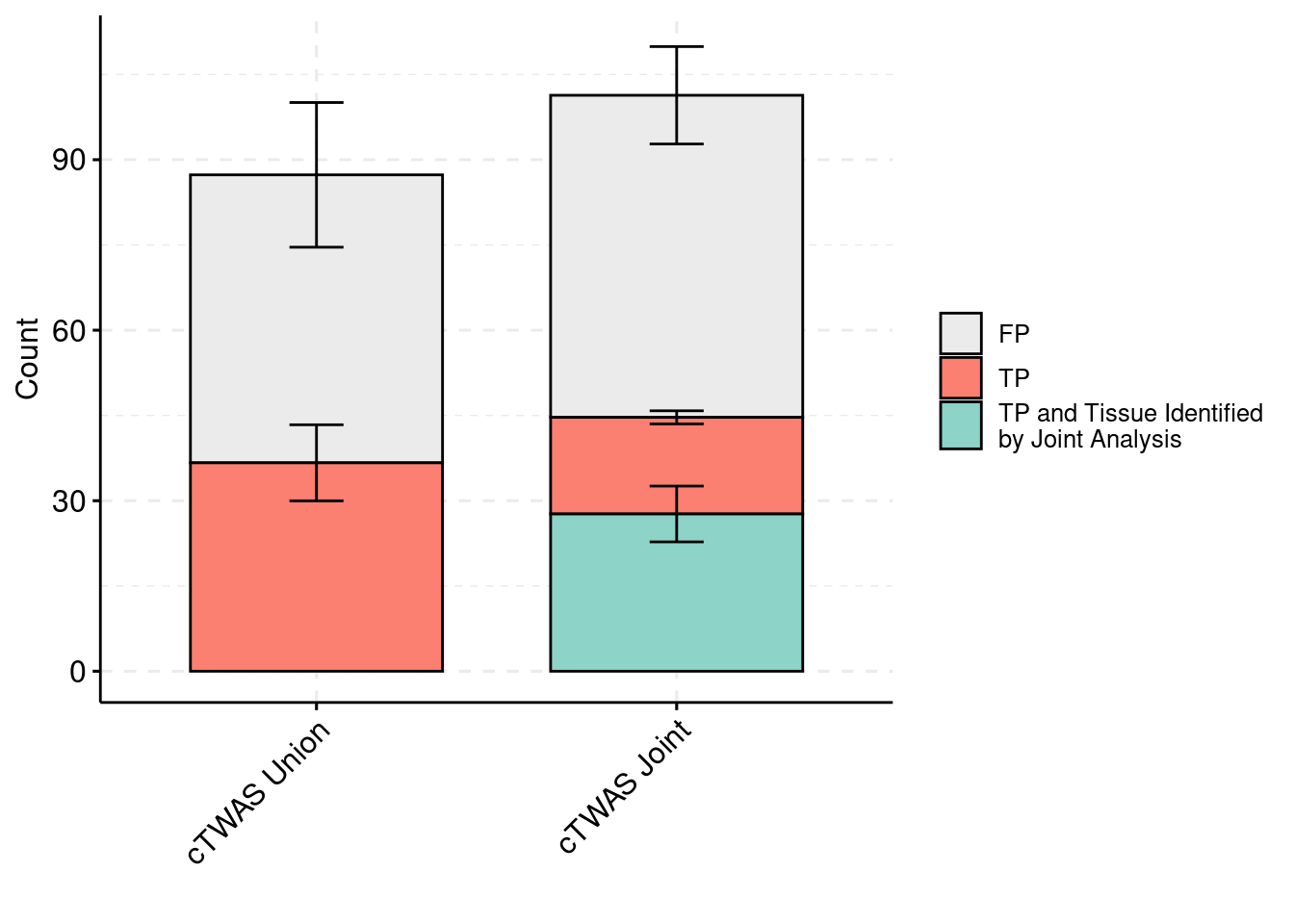
Simulation 2: Two causal tissues with equal low PVE
Separate effect size parameters
For the cTWAS analysis, each tissue had its own prior inclusion parameter and effect size parameter.
results_dir <- "/project2/xinhe/shengqian/cTWAS/cTWAS_simulation/simulation_correlated/"
runtag = "ukb-s80.45-3_corr"
configtag <- 2
simutags <- paste(2, 1:3, sep = "-")
thin <- 0.1
sample_size <- 45000
PIP_threshold <- 0.8results_df <- data.frame(simutag=as.character(),
n_causal=as.integer(),
n_causal_combined=as.integer(),
n_detected_pip=as.integer(),
n_detected_pip_in_causal=as.integer(),
n_detected_comb_pip=as.integer(),
n_detected_comb_pip_in_causal=as.integer(),
pve_snp=as.numeric(),
pve_weight1=as.numeric(),
pve_weight2=as.numeric(),
pve_weight3=as.numeric(),
prior_weight1=as.numeric(),
prior_weight2=as.numeric(),
prior_weight3=as.numeric(),
prior_var_snp=as.numeric(),
prior_var_weight1=as.numeric(),
prior_var_weight2=as.numeric(),
prior_var_weight3=as.numeric())
for (i in 1:length(simutags)){
simutag <- simutags[i]
#load genes with true simulated effect
load(paste0(results_dir, runtag, "_simu", simutag, "-pheno.Rd"))
true_genes <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$id.cgene}))
true_genes_combined <- unique(sapply(true_genes, function(x){unlist(strsplit(x, "[|]"))[1]}))
#load cTWAS results
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of causal genes
n_causal <- length(true_genes)
n_causal_combined <- length(true_genes_combined)
#number of gene+tissue combinations with cTWAS PIP > threshold
n_ctwas_genes <- sum(ctwas_gene_res$susie_pip > PIP_threshold)
#number of cTWAS genes that are causal
n_causal_detected <- sum(ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold] %in% true_genes)
#collapse gene+tissues to genes and compute combined PIP
ctwas_gene_res$gene <- sapply(ctwas_gene_res$id, function(x){unlist(strsplit(x,"[|]"))[1]})
ctwas_gene_res_combined <- aggregate(ctwas_gene_res$susie_pip, by=list(ctwas_gene_res$gene), FUN=sum)
colnames(ctwas_gene_res_combined) <- c("gene", "pip_combined")
#number of genes with combined PIP > threshold
n_ctwas_genes_combined <- sum(ctwas_gene_res_combined$pip_combined > PIP_threshold)
#number of cTWAS genes using combined PIP that are causal
n_causal_detected_combined <- sum(ctwas_gene_res_combined$gene[ctwas_gene_res_combined$pip_combined > PIP_threshold] %in% true_genes_combined)
#collect number of SNPs analyzed by cTWAS
ctwas_res_s1 <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.s1.susieIrss.txt"))
n_snps <- sum(ctwas_res_s1$type=="SNP")/thin
rm(ctwas_res_s1)
#load estimated parameters
load(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.s2.susieIrssres.Rd"))
#estimated group prior (all iterations)
estimated_group_prior_all <- group_prior_rec
estimated_group_prior_all["SNP",] <- estimated_group_prior_all["SNP",]*thin #adjust parameter to account for thin argument
#estimated group prior variance (all iterations)
estimated_group_prior_var_all <- group_prior_var_rec
#set group size
group_size <- c(table(ctwas_gene_res$type), structure(n_snps, names="SNP"))
group_size <- group_size[rownames(estimated_group_prior_all)]
#estimated group PVE (all iterations)
estimated_group_pve_all <- estimated_group_prior_var_all*estimated_group_prior_all*group_size/sample_size #check PVE calculation
results_current <- data.frame(simutag=as.character(simutag),
n_causal=as.integer(n_causal),
n_causal_combined=as.integer(n_causal_combined),
n_detected_pip=as.integer(n_ctwas_genes),
n_detected_pip_in_causal=as.integer(n_causal_detected),
n_detected_comb_pip=as.integer(n_ctwas_genes_combined),
n_detected_comb_pip_in_causal=as.integer(n_causal_detected_combined),
pve_snp=as.numeric(rev(estimated_group_pve_all["SNP",])[1]),
pve_weight1=as.numeric(rev(estimated_group_pve_all["Brain_Cerebellum_harmonized",])[1]),
pve_weight2=as.numeric(rev(estimated_group_pve_all["Brain_Hippocampus_harmonized",])[1]),
pve_weight3=as.numeric(rev(estimated_group_pve_all["Brain_Caudate_basal_ganglia_harmonized",])[1]),
prior_snp=as.numeric(rev(estimated_group_prior_var_all["SNP",])[1]),
prior_weight1=as.numeric(rev(estimated_group_prior_all["Brain_Cerebellum_harmonized",])[1]),
prior_weight2=as.numeric(rev(estimated_group_prior_all["Brain_Hippocampus_harmonized",])[1]),
prior_weight3=as.numeric(rev(estimated_group_prior_all["Brain_Caudate_basal_ganglia_harmonized",])[1]),
prior_var_snp=as.numeric(rev(estimated_group_prior_var_all["SNP",])[1]),
prior_var_weight1=as.numeric(rev(estimated_group_prior_var_all["Brain_Cerebellum_harmonized",])[1]),
prior_var_weight2=as.numeric(rev(estimated_group_prior_var_all["Brain_Hippocampus_harmonized",])[1]),
prior_var_weight3=as.numeric(rev(estimated_group_prior_var_all["Brain_Caudate_basal_ganglia_harmonized",])[1]))
results_df <- rbind(results_df, results_current)
}
#results using PIP threshold (gene+tissue)
results_df[,c("simutag", "n_causal", "n_detected_pip", "n_detected_pip_in_causal")] simutag n_causal n_detected_pip n_detected_pip_in_causal
1 2-1 66 45 9
2 2-2 77 46 6
3 2-3 65 16 3#mean percent causal using PIP > 0.8
sum(results_df$n_detected_pip_in_causal)/sum(results_df$n_detected_pip)[1] 0.1682243#results using combined PIP threshold
results_df[,c("simutag", "n_causal_combined", "n_detected_comb_pip", "n_detected_comb_pip_in_causal")] simutag n_causal_combined n_detected_comb_pip n_detected_comb_pip_in_causal
1 2-1 66 44 10
2 2-2 77 51 16
3 2-3 65 21 7#mean percent causal using combined PIP > 0.8
sum(results_df$n_detected_comb_pip_in_causal)/sum(results_df$n_detected_comb_pip)[1] 0.2844828#prior inclusion and mean prior inclusion
results_df[,c(which(colnames(results_df)=="simutag"), setdiff(grep("prior", names(results_df)), grep("prior_var", names(results_df))))] simutag prior_snp prior_weight1 prior_weight2 prior_weight3
1 2-1 8.104534 0.0017744642 0.014077209 0.004048987
2 2-2 7.339106 0.0008837711 0.001610797 0.024063238
3 2-3 8.310899 0.0030392077 0.002884784 0.015402053colMeans(results_df[,setdiff(grep("prior", names(results_df)), grep("prior_var", names(results_df)))]) prior_snp prior_weight1 prior_weight2 prior_weight3
7.918179541 0.001899148 0.006190930 0.014504760 #prior variance and mean prior variance
results_df[,c(which(colnames(results_df)=="simutag"), grep("prior_var", names(results_df)))] simutag prior_var_snp prior_var_weight1 prior_var_weight2 prior_var_weight3
1 2-1 8.104534 45.154705 5.192346 14.985668
2 2-2 7.339106 15.491046 39.302892 5.098351
3 2-3 8.310899 1.070797 8.888097 7.451467colMeans(results_df[,grep("prior_var", names(results_df))]) prior_var_snp prior_var_weight1 prior_var_weight2 prior_var_weight3
7.918180 20.572183 17.794445 9.178496 #PVE and mean PVE
results_df[,c(which(colnames(results_df)=="simutag"), grep("pve", names(results_df)))] simutag pve_snp pve_weight1 pve_weight2 pve_weight3
1 2-1 0.2596614 0.0156173316 0.012913227 0.01157039
2 2-2 0.2360318 0.0026684382 0.011184587 0.02339425
3 2-3 0.2673311 0.0006343136 0.004529776 0.02188496colMeans(results_df[,grep("pve", names(results_df))]) pve_snp pve_weight1 pve_weight2 pve_weight3
0.254341409 0.006306694 0.009542530 0.018949868 #store results for figure
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS G - Ind Var",
count=results_df$n_detected_comb_pip-results_df$n_detected_comb_pip_in_causal,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS G - Ind Var",
count=results_df$n_detected_comb_pip_in_causal,
ifcausal=T))Individual tissue analyses
For the cTWAS analysis, each tissue was analyzed individually and the results were combined.
results_dir <- "/project2/xinhe/shengqian/cTWAS/cTWAS_simulation/simulation_correlated/"
runtag = "ukb-s80.45-3_corr"
configtag <- 2
simutags <- paste(2, 1:3, sep = "-")
thin <- 0.1
sample_size <- 45000
PIP_threshold <- 0.8
results_df <- data.frame(simutag=as.character(),
n_causal_combined=as.integer(),
n_detected_weight1=as.integer(),
n_detected_in_causal_weight1=as.integer(),
n_detected_weight2=as.integer(),
n_detected_in_causal_weight2=as.integer(),
n_detected_weight3=as.integer(),
n_detected_in_causal_weight3=as.integer(),
n_detected_combined=as.integer(),
n_detected_in_causal_combined=as.integer())
for (i in 1:length(simutags)){
simutag <- simutags[i]
#load genes with true simulated effect
load(paste0(results_dir, runtag, "_simu", simutag, "-pheno.Rd"))
true_genes <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$id.cgene}))
true_genes_combined <- unique(sapply(true_genes, function(x){unlist(strsplit(x, "[|]"))[1]}))
#number of causal genes
n_causal_combined <- length(true_genes_combined)
#load cTWAS results for weight1
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight1.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight1
ctwas_genes_weight1 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight1 <- length(ctwas_genes_weight1)
n_causal_detected_weight1 <- sum(ctwas_genes_weight1 %in% true_genes_combined)
#load cTWAS results for weight2
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight2.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight1
ctwas_genes_weight2 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight2 <- length(ctwas_genes_weight2)
n_causal_detected_weight2 <- sum(ctwas_genes_weight2 %in% true_genes_combined)
#load cTWAS results for weight3
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight3.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight3
ctwas_genes_weight3 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight3 <- length(ctwas_genes_weight3)
n_causal_detected_weight3 <- sum(ctwas_genes_weight3 %in% true_genes_combined)
#combined analysis
ctwas_genes_combined <- unique(c(ctwas_genes_weight1, ctwas_genes_weight2, ctwas_genes_weight3))
n_ctwas_genes_combined <- length(ctwas_genes_combined)
n_causal_detected_combined <- sum(ctwas_genes_combined %in% true_genes_combined)
results_current <- data.frame(simutag=as.character(simutag),
n_causal_combined=as.integer(n_causal_combined),
n_detected_weight1=as.integer(n_ctwas_genes_weight1),
n_detected_in_causal_weight1=as.integer(n_causal_detected_weight1),
n_detected_weight2=as.integer(n_ctwas_genes_weight2),
n_detected_in_causal_weight2=as.integer(n_causal_detected_weight2),
n_detected_weight3=as.integer(n_ctwas_genes_weight3),
n_detected_in_causal_weight3=as.integer(n_causal_detected_weight3),
n_detected_combined=as.integer(n_ctwas_genes_combined),
n_detected_in_causal_combined=as.integer(n_causal_detected_combined))
results_df <- rbind(results_df, results_current)
}
#results using weight1
results_df[,c("simutag", colnames(results_df)[grep("weight1", colnames(results_df))])] simutag n_detected_weight1 n_detected_in_causal_weight1
1 2-1 32 8
2 2-2 22 3
3 2-3 19 3#mean percent causal using PIP > 0.8 for weight1
sum(results_df$n_detected_in_causal_weight1)/sum(results_df$n_detected_weight1)[1] 0.1917808#results using weight2
results_df[,c("simutag", colnames(results_df)[grep("weight2", colnames(results_df))])] simutag n_detected_weight2 n_detected_in_causal_weight2
1 2-1 34 4
2 2-2 30 12
3 2-3 24 4#mean percent causal using PIP > 0.8 for weight1
sum(results_df$n_detected_in_causal_weight2)/sum(results_df$n_detected_weight2)[1] 0.2272727#results using weight3
results_df[,c("simutag", colnames(results_df)[grep("weight3", colnames(results_df))])] simutag n_detected_weight3 n_detected_in_causal_weight3
1 2-1 29 3
2 2-2 0 0
3 2-3 33 9#mean percent causal using PIP > 0.8 for weight3
sum(results_df$n_detected_in_causal_weight3)/sum(results_df$n_detected_weight3)[1] 0.1935484#results using combined analysis
results_df[,c("simutag", colnames(results_df)[grep("combined", colnames(results_df))])] simutag n_causal_combined n_detected_combined n_detected_in_causal_combined
1 2-1 66 72 12
2 2-2 77 46 13
3 2-3 65 63 12#mean percent causal using PIP > 0.8 for weight3
sum(results_df$n_detected_in_causal_combined)/sum(results_df$n_detected_combined)[1] 0.2044199#store results for figure
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight1",
count=results_df$n_detected_weight1-results_df$n_detected_in_causal_weight1,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight1",
count=results_df$n_detected_in_causal_weight1,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight2",
count=results_df$n_detected_weight2-results_df$n_detected_in_causal_weight2,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight2",
count=results_df$n_detected_in_causal_weight2,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight3",
count=results_df$n_detected_weight3-results_df$n_detected_in_causal_weight3,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight3",
count=results_df$n_detected_in_causal_weight3,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Union",
count=results_df$n_detected_combined-results_df$n_detected_in_causal_combined,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Union",
count=results_df$n_detected_in_causal_combined,
ifcausal=T))Figure
For the cTWAS analysis, each tissue was analyzed individually and the results were combined.
plot_df$method <- factor(plot_df$method, levels=c("TWAS", "cTWAS G+T", "cTWAS G", "cTWAS G - Ind Var", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3"))
library(ggpubr)
colset = c("#ebebeb", "#fb8072")
ggbarplot(plot_df,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "none",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1))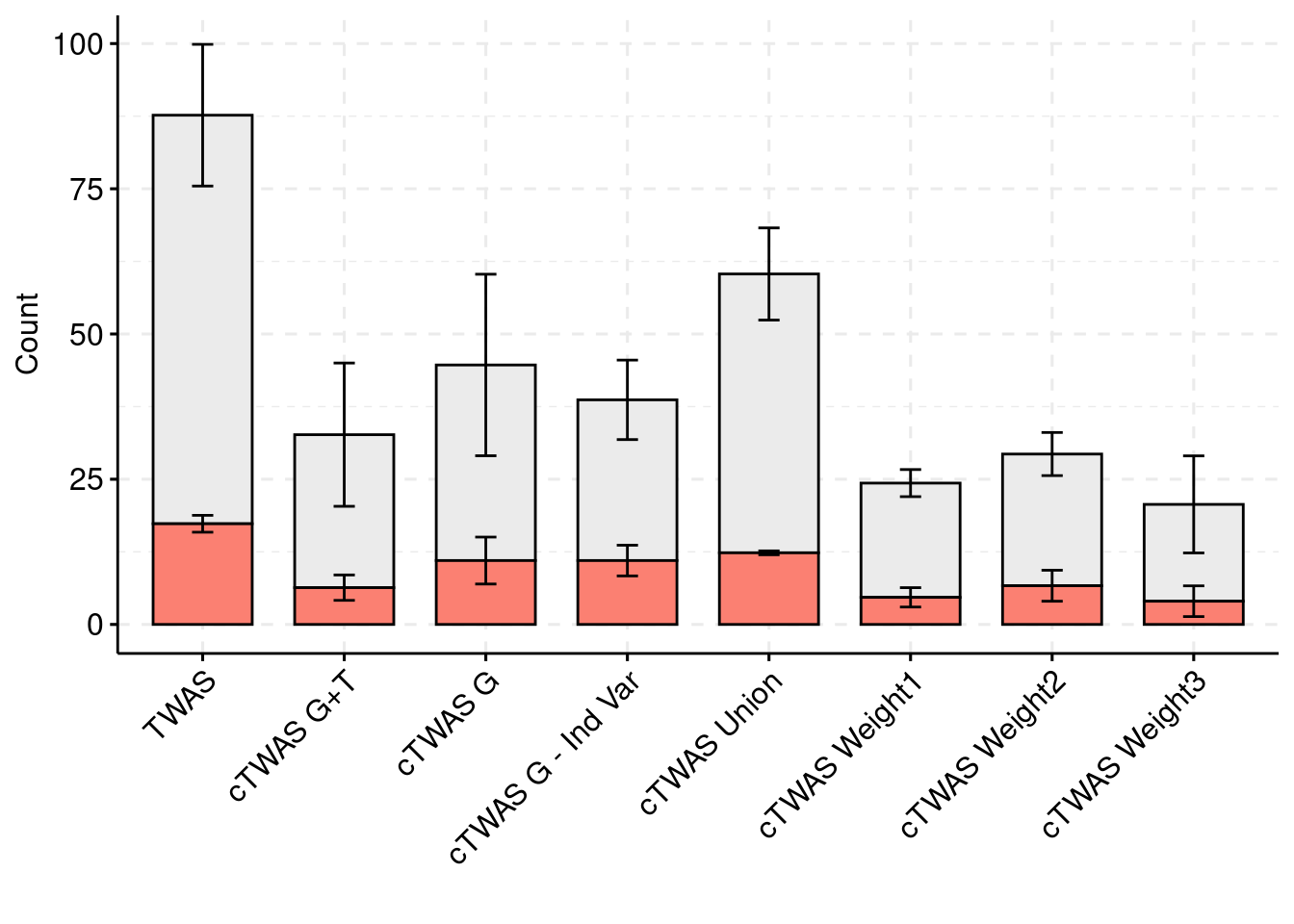
plot_df_2 <- plot_df
plot_df_2$method %in% c("TWAS", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3") [1] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE
[13] TRUE TRUE TRUE TRUE TRUE TRUE FALSE FALSE FALSE FALSE FALSE FALSE
[25] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
[37] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUEplot_df_2$ifcausal <- as.character(plot_df_2$ifcausal+1)
plot_df_2$count[plot_df_2$method=="cTWAS G" & plot_df_2$ifcausal=="2"] <- plot_df_2$count[plot_df_2$method=="cTWAS G" & plot_df_2$ifcausal=="2"] - plot_df_2$count[plot_df_2$method=="cTWAS G+T" & plot_df_2$ifcausal=="2"]
plot_df_2 <- rbind(plot_df_2, data.frame(simutag=simutags,
method="cTWAS G",
count=plot_df_2$count[plot_df_2$method=="cTWAS G+T" & plot_df_2$ifcausal=="2"],
ifcausal="3"))
plot_df_3 <- plot_df_2
plot_df_2 <- plot_df_2[plot_df_2$method %in% c("TWAS", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3"),]
plot_df_2$method <- droplevels(plot_df_2$method)
levels(plot_df_2$method) <- c("TWAS", "cTWAS Union", "cTWAS Primary", "cTWAS Secondary", "cTWAS Null")
plot_df_2$ifcausal[plot_df_2$ifcausal=="1"] <- "FP"
plot_df_2$ifcausal[plot_df_2$ifcausal=="2"] <- "TP"
colset = c("#ebebeb", "#fb8072", "#8dd3c7")
ggbarplot(plot_df_2,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "right",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1)) + guides(fill=guide_legend(title=""))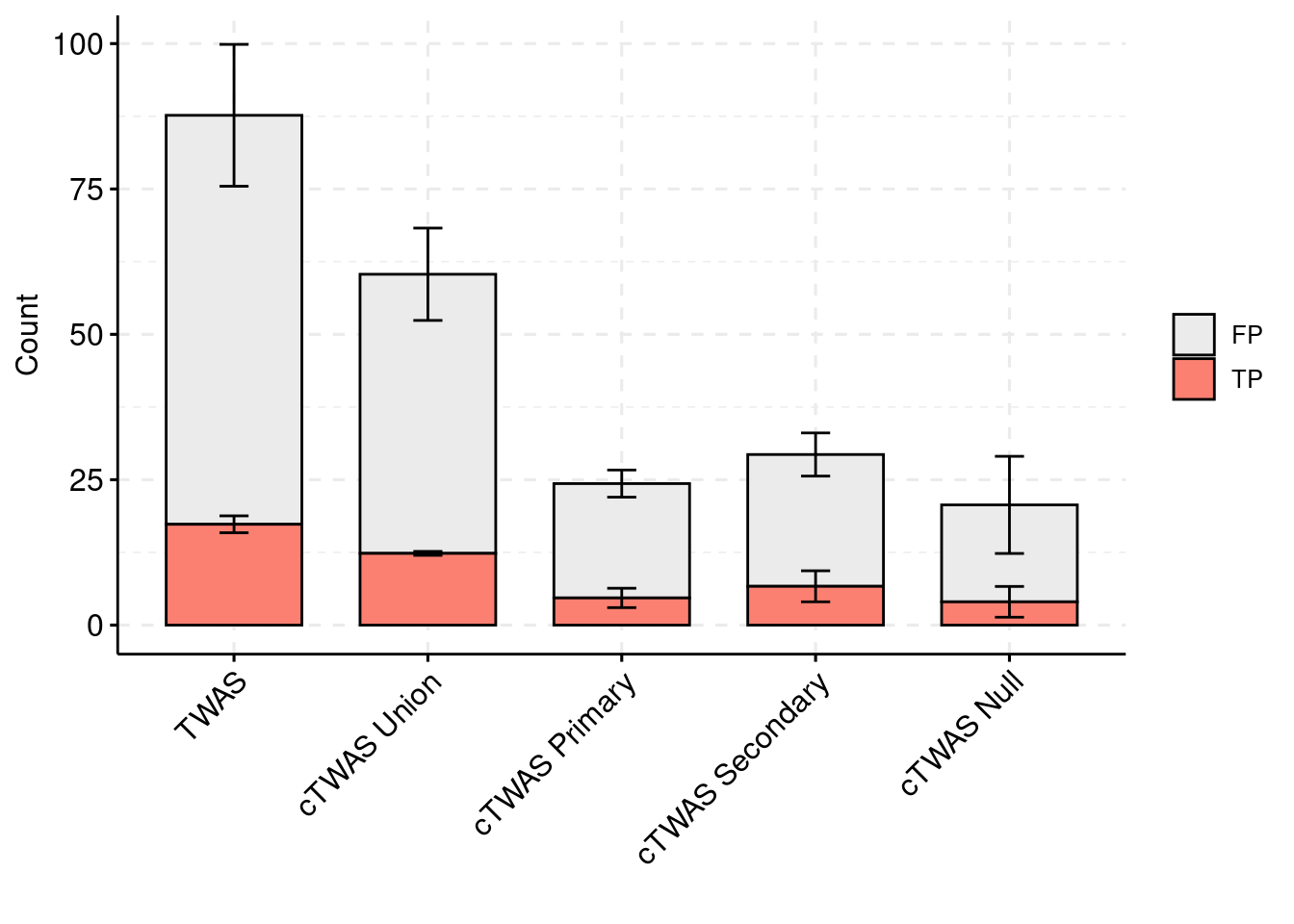
plot_df_3 <- plot_df_3[plot_df_3$method %in% c("cTWAS G", "cTWAS Union"),]
plot_df_3$method <- droplevels(plot_df_3$method)
levels(plot_df_3$method) <- c("cTWAS Joint", "cTWAS Union")
plot_df_3$method <- relevel(plot_df_3$method, "cTWAS Union")
plot_df_3$ifcausal[plot_df_3$ifcausal=="1"] <- "FP"
plot_df_3$ifcausal[plot_df_3$ifcausal=="2"] <- "TP"
plot_df_3$ifcausal[plot_df_3$ifcausal=="3"] <- "TP and Tissue Identified\nby Joint Analysis"
ggbarplot(plot_df_3,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "right",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1)) + guides(fill=guide_legend(title=""))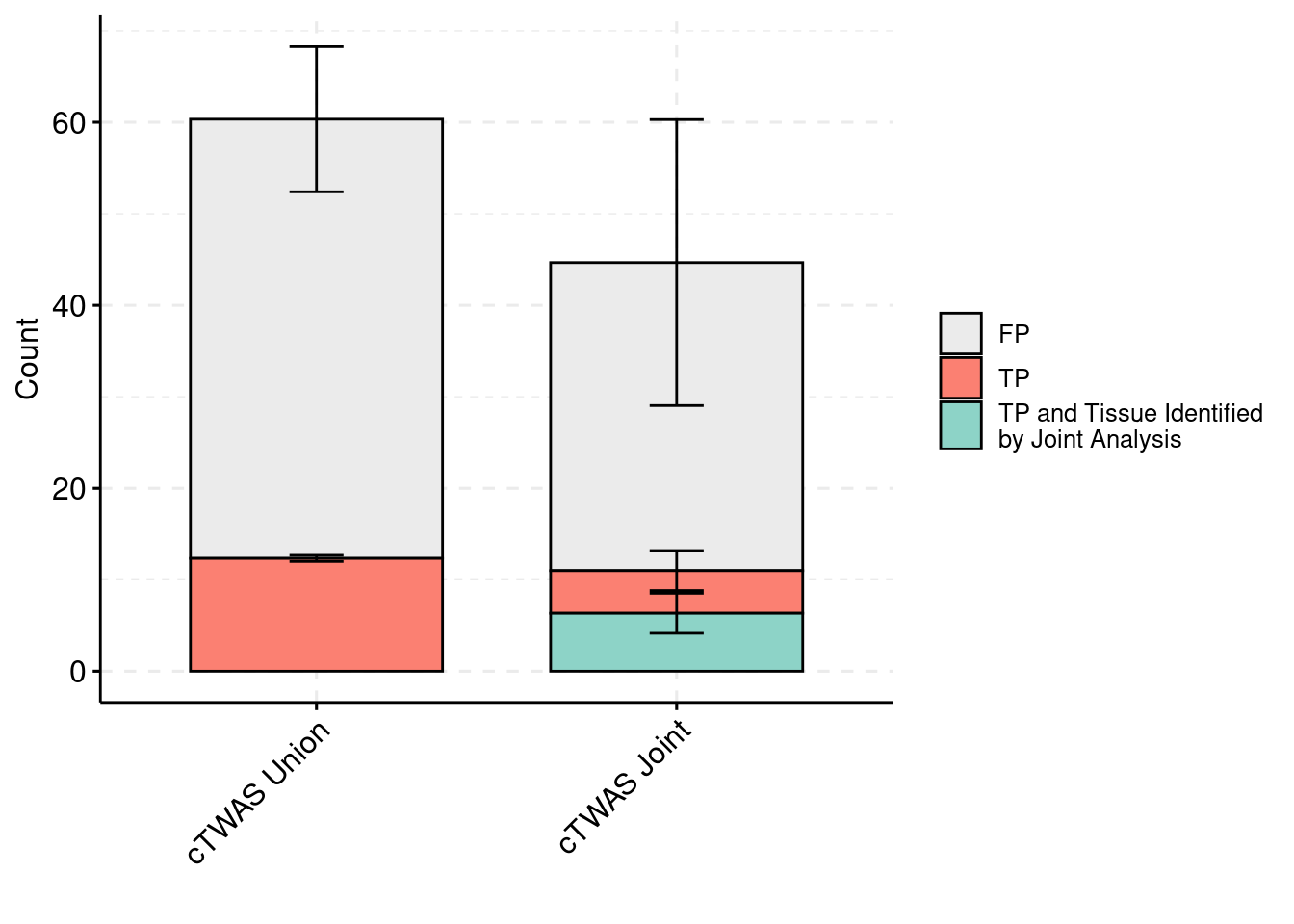
Simulation 3: Three causal tissues with unequal PVE
Separate effect size parameters
For the cTWAS analysis, each tissue had its own prior inclusion parameter and effect size parameter.
results_dir <- "/project2/xinhe/shengqian/cTWAS/cTWAS_simulation/simulation_correlated/"
runtag = "ukb-s80.45-3_corr"
configtag <- 2
simutags <- paste(3, 1:3, sep = "-")
thin <- 0.1
sample_size <- 45000
PIP_threshold <- 0.8
results_df <- data.frame(simutag=as.character(),
n_causal=as.integer(),
n_causal_combined=as.integer(),
n_detected_pip=as.integer(),
n_detected_pip_in_causal=as.integer(),
n_detected_comb_pip=as.integer(),
n_detected_comb_pip_in_causal=as.integer(),
pve_snp=as.numeric(),
pve_weight1=as.numeric(),
pve_weight2=as.numeric(),
pve_weight3=as.numeric(),
prior_weight1=as.numeric(),
prior_weight2=as.numeric(),
prior_weight3=as.numeric(),
prior_var_snp=as.numeric(),
prior_var_weight1=as.numeric(),
prior_var_weight2=as.numeric(),
prior_var_weight3=as.numeric())
for (i in 1:length(simutags)){
simutag <- simutags[i]
#load genes with true simulated effect
load(paste0(results_dir, runtag, "_simu", simutag, "-pheno.Rd"))
true_genes <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$id.cgene}))
true_genes_combined <- unique(sapply(true_genes, function(x){unlist(strsplit(x, "[|]"))[1]}))
#load cTWAS results
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of causal genes
n_causal <- length(true_genes)
n_causal_combined <- length(true_genes_combined)
#number of gene+tissue combinations with cTWAS PIP > threshold
n_ctwas_genes <- sum(ctwas_gene_res$susie_pip > PIP_threshold)
#number of cTWAS genes that are causal
n_causal_detected <- sum(ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold] %in% true_genes)
#collapse gene+tissues to genes and compute combined PIP
ctwas_gene_res$gene <- sapply(ctwas_gene_res$id, function(x){unlist(strsplit(x,"[|]"))[1]})
ctwas_gene_res_combined <- aggregate(ctwas_gene_res$susie_pip, by=list(ctwas_gene_res$gene), FUN=sum)
colnames(ctwas_gene_res_combined) <- c("gene", "pip_combined")
#number of genes with combined PIP > threshold
n_ctwas_genes_combined <- sum(ctwas_gene_res_combined$pip_combined > PIP_threshold)
#number of cTWAS genes using combined PIP that are causal
n_causal_detected_combined <- sum(ctwas_gene_res_combined$gene[ctwas_gene_res_combined$pip_combined > PIP_threshold] %in% true_genes_combined)
#collect number of SNPs analyzed by cTWAS
ctwas_res_s1 <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.s1.susieIrss.txt"))
n_snps <- sum(ctwas_res_s1$type=="SNP")/thin
rm(ctwas_res_s1)
#load estimated parameters
load(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.s2.susieIrssres.Rd"))
#estimated group prior (all iterations)
estimated_group_prior_all <- group_prior_rec
estimated_group_prior_all["SNP",] <- estimated_group_prior_all["SNP",]*thin #adjust parameter to account for thin argument
#estimated group prior variance (all iterations)
estimated_group_prior_var_all <- group_prior_var_rec
#set group size
group_size <- c(table(ctwas_gene_res$type), structure(n_snps, names="SNP"))
group_size <- group_size[rownames(estimated_group_prior_all)]
#estimated group PVE (all iterations)
estimated_group_pve_all <- estimated_group_prior_var_all*estimated_group_prior_all*group_size/sample_size #check PVE calculation
results_current <- data.frame(simutag=as.character(simutag),
n_causal=as.integer(n_causal),
n_causal_combined=as.integer(n_causal_combined),
n_detected_pip=as.integer(n_ctwas_genes),
n_detected_pip_in_causal=as.integer(n_causal_detected),
n_detected_comb_pip=as.integer(n_ctwas_genes_combined),
n_detected_comb_pip_in_causal=as.integer(n_causal_detected_combined),
pve_snp=as.numeric(rev(estimated_group_pve_all["SNP",])[1]),
pve_weight1=as.numeric(rev(estimated_group_pve_all["Brain_Cerebellum_harmonized",])[1]),
pve_weight2=as.numeric(rev(estimated_group_pve_all["Brain_Hippocampus_harmonized",])[1]),
pve_weight3=as.numeric(rev(estimated_group_pve_all["Brain_Caudate_basal_ganglia_harmonized",])[1]),
prior_snp=as.numeric(rev(estimated_group_prior_var_all["SNP",])[1]),
prior_weight1=as.numeric(rev(estimated_group_prior_all["Brain_Cerebellum_harmonized",])[1]),
prior_weight2=as.numeric(rev(estimated_group_prior_all["Brain_Hippocampus_harmonized",])[1]),
prior_weight3=as.numeric(rev(estimated_group_prior_all["Brain_Caudate_basal_ganglia_harmonized",])[1]),
prior_var_snp=as.numeric(rev(estimated_group_prior_var_all["SNP",])[1]),
prior_var_weight1=as.numeric(rev(estimated_group_prior_var_all["Brain_Cerebellum_harmonized",])[1]),
prior_var_weight2=as.numeric(rev(estimated_group_prior_var_all["Brain_Hippocampus_harmonized",])[1]),
prior_var_weight3=as.numeric(rev(estimated_group_prior_var_all["Brain_Caudate_basal_ganglia_harmonized",])[1]))
results_df <- rbind(results_df, results_current)
}
#results using PIP threshold (gene+tissue)
results_df[,c("simutag", "n_causal", "n_detected_pip", "n_detected_pip_in_causal")] simutag n_causal n_detected_pip n_detected_pip_in_causal
1 3-1 137 82 25
2 3-2 158 57 16
3 3-3 135 79 12#mean percent causal using PIP > 0.8
sum(results_df$n_detected_pip_in_causal)/sum(results_df$n_detected_pip)[1] 0.2431193#results using combined PIP threshold
results_df[,c("simutag", "n_causal_combined", "n_detected_comb_pip", "n_detected_comb_pip_in_causal")] simutag n_causal_combined n_detected_comb_pip n_detected_comb_pip_in_causal
1 3-1 137 79 30
2 3-2 156 59 25
3 3-3 134 84 21#mean percent causal using combined PIP > 0.8
sum(results_df$n_detected_comb_pip_in_causal)/sum(results_df$n_detected_comb_pip)[1] 0.3423423#prior inclusion and mean prior inclusion
results_df[,c(which(colnames(results_df)=="simutag"), setdiff(grep("prior", names(results_df)), grep("prior_var", names(results_df))))] simutag prior_snp prior_weight1 prior_weight2 prior_weight3
1 3-1 7.694010 0.009496299 0.015700714 0.0005218101
2 3-2 8.968999 0.010896212 0.002214315 0.0065810326
3 3-3 8.327279 0.011588585 0.003198182 0.0106307116colMeans(results_df[,setdiff(grep("prior", names(results_df)), grep("prior_var", names(results_df)))]) prior_snp prior_weight1 prior_weight2 prior_weight3
8.330095836 0.010660365 0.007037737 0.005911185 #prior variance and mean prior variance
results_df[,c(which(colnames(results_df)=="simutag"), grep("prior_var", names(results_df)))] simutag prior_var_snp prior_var_weight1 prior_var_weight2 prior_var_weight3
1 3-1 7.694010 20.04747 11.83834 102.068952
2 3-2 8.968999 19.77378 1.67682 16.497945
3 3-3 8.327279 15.43960 31.66265 3.725893colMeans(results_df[,grep("prior_var", names(results_df))]) prior_var_snp prior_var_weight1 prior_var_weight2 prior_var_weight3
8.330096 18.420282 15.059270 40.764263 #PVE and mean PVE
results_df[,c(which(colnames(results_df)=="simutag"), grep("pve", names(results_df)))] simutag pve_snp pve_weight1 pve_weight2 pve_weight3
1 3-1 0.2386009 0.03710654 0.0328371066 0.010156208
2 3-2 0.3126840 0.04199542 0.0006559645 0.020703762
3 3-3 0.2762783 0.03487410 0.0178897832 0.007552976colMeans(results_df[,grep("pve", names(results_df))]) pve_snp pve_weight1 pve_weight2 pve_weight3
0.27585440 0.03799202 0.01712762 0.01280432 #store results for figure
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS G - Ind Var",
count=results_df$n_detected_comb_pip-results_df$n_detected_comb_pip_in_causal,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS G - Ind Var",
count=results_df$n_detected_comb_pip_in_causal,
ifcausal=T))Individual tissue analyses
For the cTWAS analysis, each tissue was analyzed individually and the results were combined.
results_dir <- "/project2/xinhe/shengqian/cTWAS/cTWAS_simulation/simulation_correlated/"
runtag = "ukb-s80.45-3_corr"
configtag <- 2
simutags <- paste(3, 1:3, sep = "-")
thin <- 0.1
sample_size <- 45000
PIP_threshold <- 0.8
results_df <- data.frame(simutag=as.character(),
n_causal_combined=as.integer(),
n_detected_weight1=as.integer(),
n_detected_in_causal_weight1=as.integer(),
n_detected_weight2=as.integer(),
n_detected_in_causal_weight2=as.integer(),
n_detected_weight3=as.integer(),
n_detected_in_causal_weight3=as.integer(),
n_detected_combined=as.integer(),
n_detected_in_causal_combined=as.integer())
for (i in 1:length(simutags)){
simutag <- simutags[i]
#load genes with true simulated effect
load(paste0(results_dir, runtag, "_simu", simutag, "-pheno.Rd"))
true_genes <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$id.cgene}))
true_genes_combined <- unique(sapply(true_genes, function(x){unlist(strsplit(x, "[|]"))[1]}))
#number of causal genes
n_causal_combined <- length(true_genes_combined)
#load cTWAS results for weight1
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight1.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight1
ctwas_genes_weight1 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight1 <- length(ctwas_genes_weight1)
n_causal_detected_weight1 <- sum(ctwas_genes_weight1 %in% true_genes_combined)
#load cTWAS results for weight2
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight2.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight1
ctwas_genes_weight2 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight2 <- length(ctwas_genes_weight2)
n_causal_detected_weight2 <- sum(ctwas_genes_weight2 %in% true_genes_combined)
#load cTWAS results for weight3
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight3.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight3
ctwas_genes_weight3 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight3 <- length(ctwas_genes_weight3)
n_causal_detected_weight3 <- sum(ctwas_genes_weight3 %in% true_genes_combined)
#combined analysis
ctwas_genes_combined <- unique(c(ctwas_genes_weight1, ctwas_genes_weight2, ctwas_genes_weight3))
n_ctwas_genes_combined <- length(ctwas_genes_combined)
n_causal_detected_combined <- sum(ctwas_genes_combined %in% true_genes_combined)
results_current <- data.frame(simutag=as.character(simutag),
n_causal_combined=as.integer(n_causal_combined),
n_detected_weight1=as.integer(n_ctwas_genes_weight1),
n_detected_in_causal_weight1=as.integer(n_causal_detected_weight1),
n_detected_weight2=as.integer(n_ctwas_genes_weight2),
n_detected_in_causal_weight2=as.integer(n_causal_detected_weight2),
n_detected_weight3=as.integer(n_ctwas_genes_weight3),
n_detected_in_causal_weight3=as.integer(n_causal_detected_weight3),
n_detected_combined=as.integer(n_ctwas_genes_combined),
n_detected_in_causal_combined=as.integer(n_causal_detected_combined))
results_df <- rbind(results_df, results_current)
}
#results using weight1
results_df[,c("simutag", colnames(results_df)[grep("weight1", colnames(results_df))])] simutag n_detected_weight1 n_detected_in_causal_weight1
1 3-1 27 10
2 3-2 29 15
3 3-3 52 11#mean percent causal using PIP > 0.8 for weight1
sum(results_df$n_detected_in_causal_weight1)/sum(results_df$n_detected_weight1)[1] 0.3333333#results using weight2
results_df[,c("simutag", colnames(results_df)[grep("weight2", colnames(results_df))])] simutag n_detected_weight2 n_detected_in_causal_weight2
1 3-1 40 15
2 3-2 32 11
3 3-3 47 13#mean percent causal using PIP > 0.8 for weight1
sum(results_df$n_detected_in_causal_weight2)/sum(results_df$n_detected_weight2)[1] 0.3277311#results using weight3
results_df[,c("simutag", colnames(results_df)[grep("weight3", colnames(results_df))])] simutag n_detected_weight3 n_detected_in_causal_weight3
1 3-1 35 10
2 3-2 33 10
3 3-3 24 7#mean percent causal using PIP > 0.8 for weight3
sum(results_df$n_detected_in_causal_weight3)/sum(results_df$n_detected_weight3)[1] 0.2934783#results using combined analysis
results_df[,c("simutag", colnames(results_df)[grep("combined", colnames(results_df))])] simutag n_causal_combined n_detected_combined n_detected_in_causal_combined
1 3-1 137 76 26
2 3-2 156 71 27
3 3-3 134 96 22#mean percent causal using PIP > 0.8 for weight3
sum(results_df$n_detected_in_causal_combined)/sum(results_df$n_detected_combined)[1] 0.308642#store results for figure
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight1",
count=results_df$n_detected_weight1-results_df$n_detected_in_causal_weight1,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight1",
count=results_df$n_detected_in_causal_weight1,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight2",
count=results_df$n_detected_weight2-results_df$n_detected_in_causal_weight2,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight2",
count=results_df$n_detected_in_causal_weight2,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight3",
count=results_df$n_detected_weight3-results_df$n_detected_in_causal_weight3,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight3",
count=results_df$n_detected_in_causal_weight3,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Union",
count=results_df$n_detected_combined-results_df$n_detected_in_causal_combined,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Union",
count=results_df$n_detected_in_causal_combined,
ifcausal=T))Figure
For the cTWAS analysis, each tissue was analyzed individually and the results were combined.
plot_df$method <- factor(plot_df$method, levels=c("TWAS", "cTWAS G+T", "cTWAS G", "cTWAS G - Ind Var", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3"))
library(ggpubr)
colset = c("#ebebeb", "#fb8072")
ggbarplot(plot_df,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "none",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1))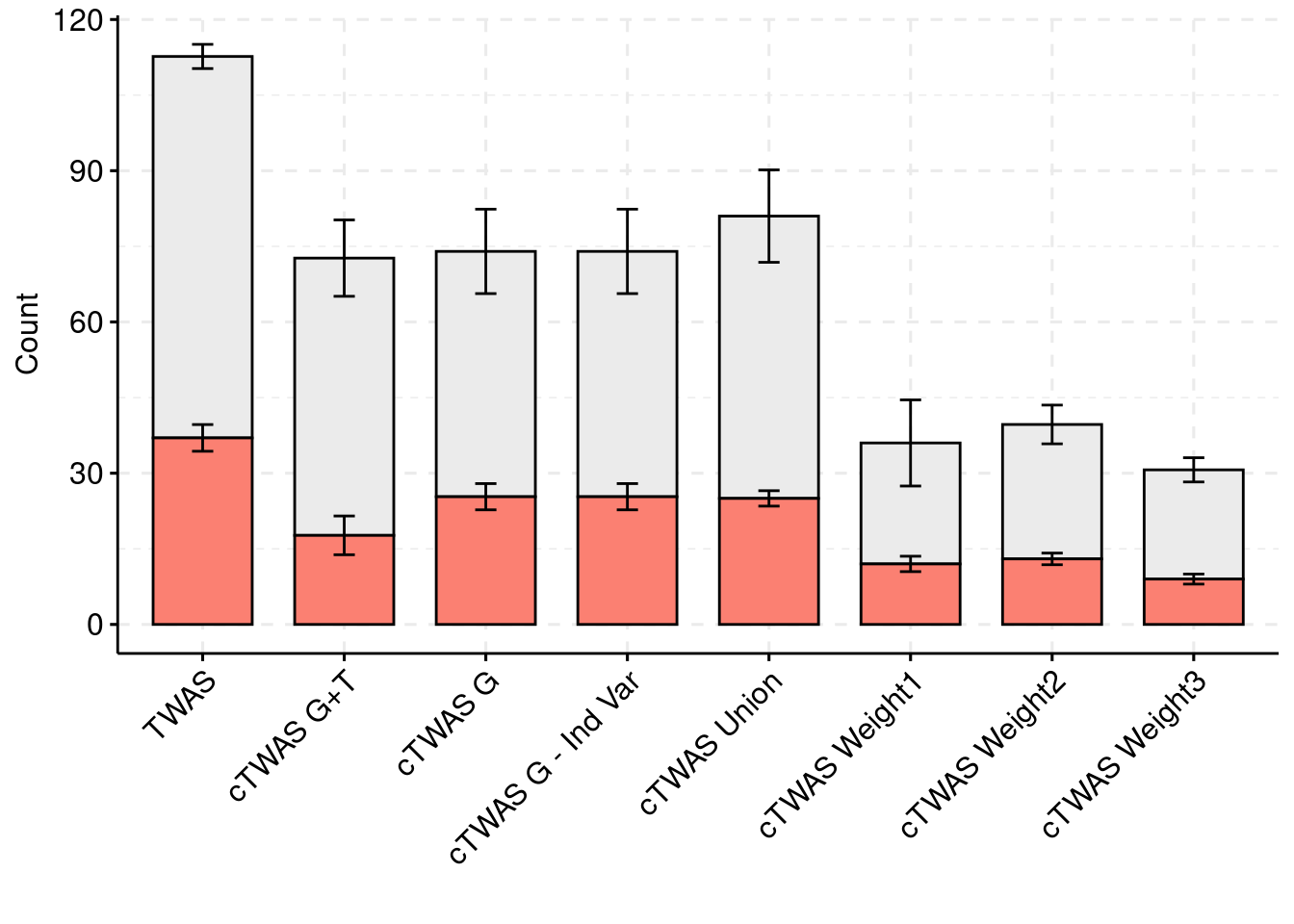
plot_df_2 <- plot_df
plot_df_2$method %in% c("TWAS", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3") [1] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE
[13] TRUE TRUE TRUE TRUE TRUE TRUE FALSE FALSE FALSE FALSE FALSE FALSE
[25] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
[37] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUEplot_df_2$ifcausal <- as.character(plot_df_2$ifcausal+1)
plot_df_2$count[plot_df_2$method=="cTWAS G" & plot_df_2$ifcausal=="2"] <- plot_df_2$count[plot_df_2$method=="cTWAS G" & plot_df_2$ifcausal=="2"] - plot_df_2$count[plot_df_2$method=="cTWAS G+T" & plot_df_2$ifcausal=="2"]
plot_df_2 <- rbind(plot_df_2, data.frame(simutag=simutags,
method="cTWAS G",
count=plot_df_2$count[plot_df_2$method=="cTWAS G+T" & plot_df_2$ifcausal=="2"],
ifcausal="3"))
plot_df_3 <- plot_df_2
plot_df_2 <- plot_df_2[plot_df_2$method %in% c("TWAS", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3"),]
plot_df_2$method <- droplevels(plot_df_2$method)
levels(plot_df_2$method) <- c("TWAS", "cTWAS Union", "cTWAS Primary", "cTWAS Secondary", "cTWAS Null")
plot_df_2$ifcausal[plot_df_2$ifcausal=="1"] <- "FP"
plot_df_2$ifcausal[plot_df_2$ifcausal=="2"] <- "TP"
colset = c("#ebebeb", "#fb8072", "#8dd3c7")
ggbarplot(plot_df_2,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "right",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1)) + guides(fill=guide_legend(title=""))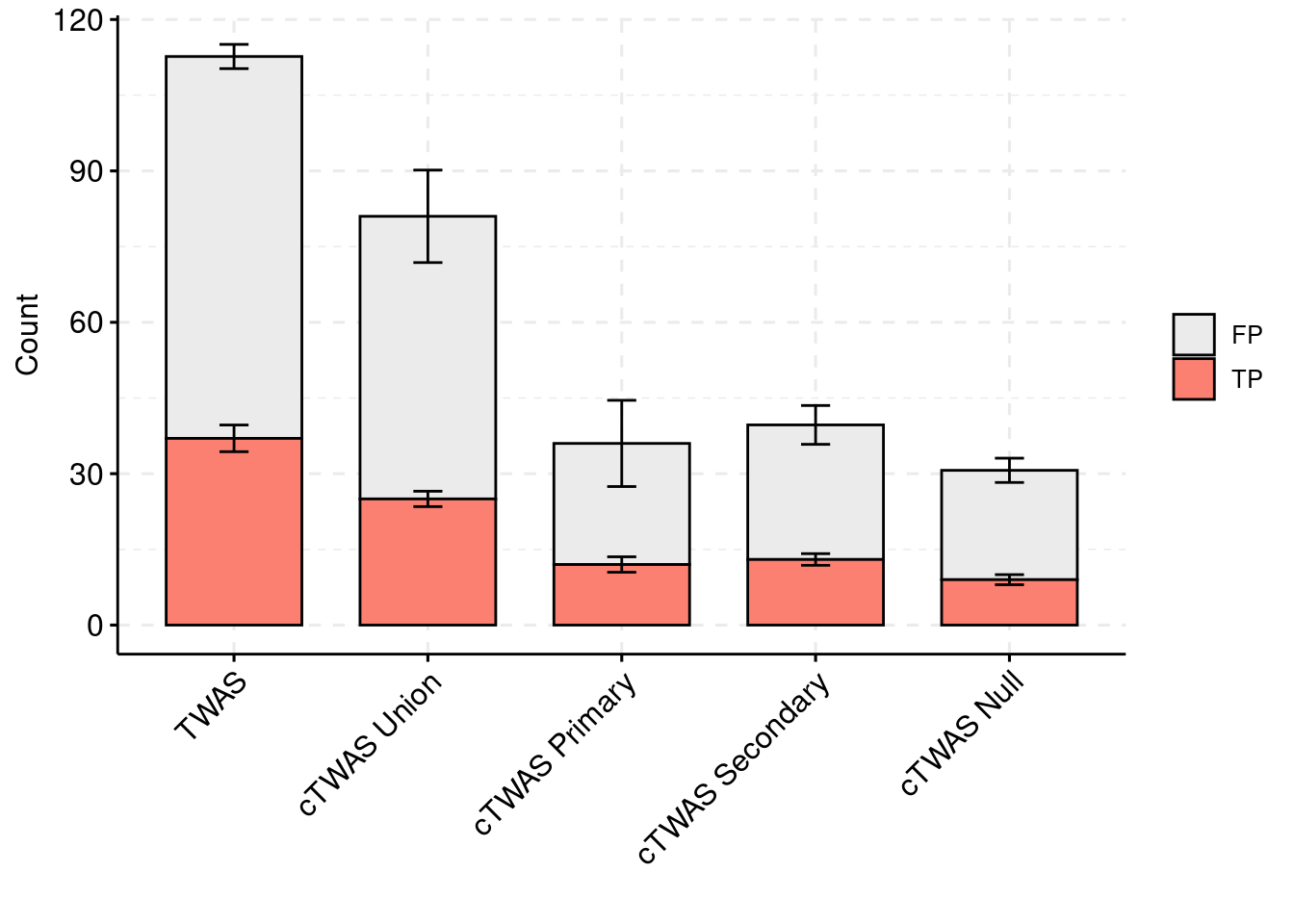
plot_df_3 <- plot_df_3[plot_df_3$method %in% c("cTWAS G", "cTWAS Union"),]
plot_df_3$method <- droplevels(plot_df_3$method)
levels(plot_df_3$method) <- c("cTWAS Joint", "cTWAS Union")
plot_df_3$method <- relevel(plot_df_3$method, "cTWAS Union")
plot_df_3$ifcausal[plot_df_3$ifcausal=="1"] <- "FP"
plot_df_3$ifcausal[plot_df_3$ifcausal=="2"] <- "TP"
plot_df_3$ifcausal[plot_df_3$ifcausal=="3"] <- "TP and Tissue Identified\nby Joint Analysis"
ggbarplot(plot_df_3,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "right",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1)) + guides(fill=guide_legend(title=""))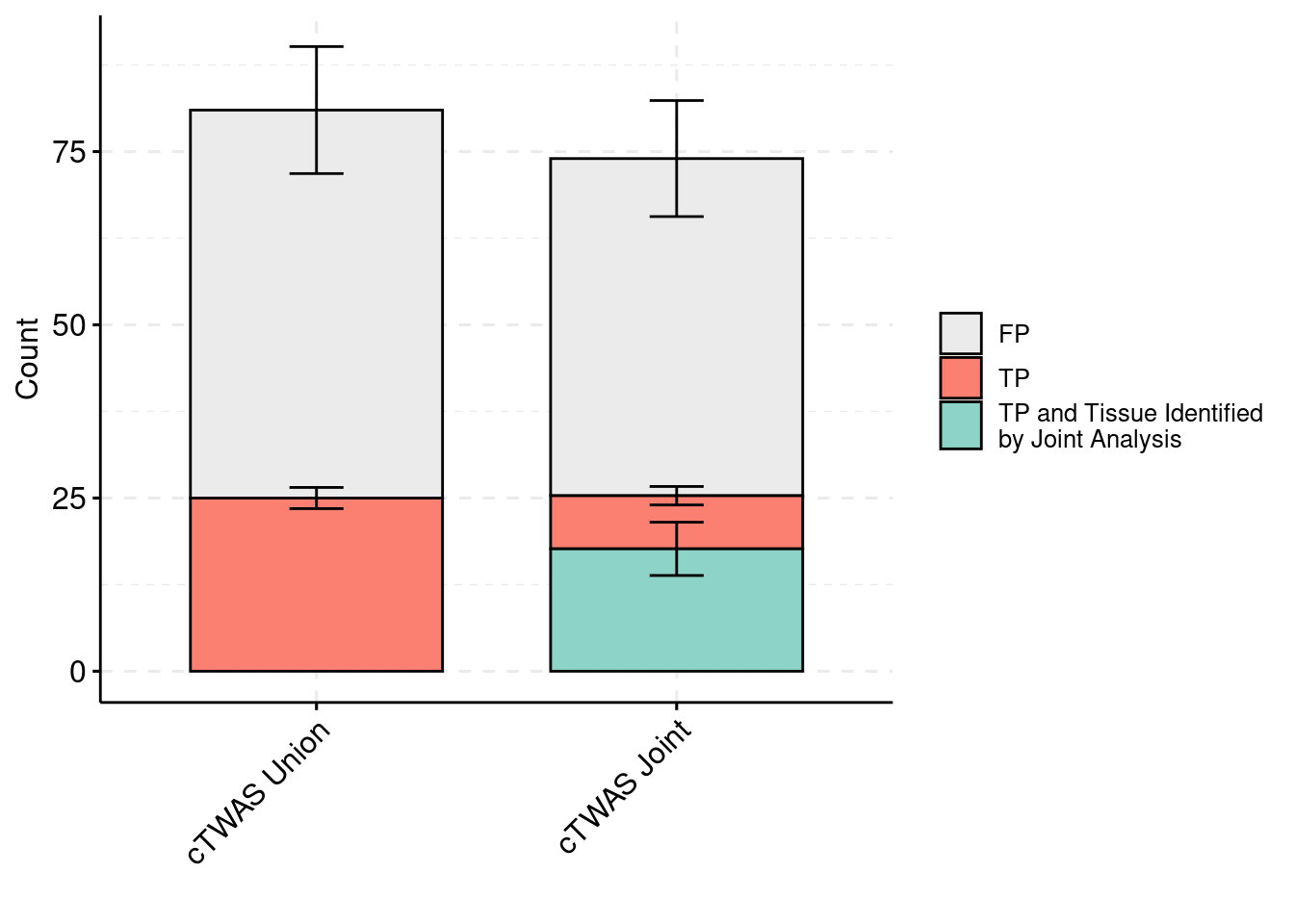
Simulation 4: Two causal tissues with unequal PVE
Separate effect size parameters
For the cTWAS analysis, each tissue had its own prior inclusion parameter end effect size parameter.
results_dir <- "/project2/xinhe/shengqian/cTWAS/cTWAS_simulation/simulation_correlated/"
runtag = "ukb-s80.45-3_corr"
configtag <- 2
simutags <- paste(4, 1:3, sep = "-")
thin <- 0.1
sample_size <- 45000
PIP_threshold <- 0.8
results_df <- data.frame(simutag=as.character(),
n_causal=as.integer(),
n_causal_combined=as.integer(),
n_detected_pip=as.integer(),
n_detected_pip_in_causal=as.integer(),
n_detected_comb_pip=as.integer(),
n_detected_comb_pip_in_causal=as.integer(),
pve_snp=as.numeric(),
pve_weight1=as.numeric(),
pve_weight2=as.numeric(),
pve_weight3=as.numeric(),
prior_weight1=as.numeric(),
prior_weight2=as.numeric(),
prior_weight3=as.numeric(),
prior_var_snp=as.numeric(),
prior_var_weight1=as.numeric(),
prior_var_weight2=as.numeric(),
prior_var_weight3=as.numeric())
for (i in 1:length(simutags)){
simutag <- simutags[i]
#load genes with true simulated effect
load(paste0(results_dir, runtag, "_simu", simutag, "-pheno.Rd"))
true_genes <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$id.cgene}))
true_genes_combined <- unique(sapply(true_genes, function(x){unlist(strsplit(x, "[|]"))[1]}))
#load cTWAS results
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of causal genes
n_causal <- length(true_genes)
n_causal_combined <- length(true_genes_combined)
#number of gene+tissue combinations with cTWAS PIP > threshold
n_ctwas_genes <- sum(ctwas_gene_res$susie_pip > PIP_threshold)
#number of cTWAS genes that are causal
n_causal_detected <- sum(ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold] %in% true_genes)
#collapse gene+tissues to genes and compute combined PIP
ctwas_gene_res$gene <- sapply(ctwas_gene_res$id, function(x){unlist(strsplit(x,"[|]"))[1]})
ctwas_gene_res_combined <- aggregate(ctwas_gene_res$susie_pip, by=list(ctwas_gene_res$gene), FUN=sum)
colnames(ctwas_gene_res_combined) <- c("gene", "pip_combined")
#number of genes with combined PIP > threshold
n_ctwas_genes_combined <- sum(ctwas_gene_res_combined$pip_combined > PIP_threshold)
#number of cTWAS genes using combined PIP that are causal
n_causal_detected_combined <- sum(ctwas_gene_res_combined$gene[ctwas_gene_res_combined$pip_combined > PIP_threshold] %in% true_genes_combined)
#collect number of SNPs analyzed by cTWAS
ctwas_res_s1 <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.s1.susieIrss.txt"))
n_snps <- sum(ctwas_res_s1$type=="SNP")/thin
rm(ctwas_res_s1)
#load estimated parameters
load(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR.s2.susieIrssres.Rd"))
#estimated group prior (all iterations)
estimated_group_prior_all <- group_prior_rec
estimated_group_prior_all["SNP",] <- estimated_group_prior_all["SNP",]*thin #adjust parameter to account for thin argument
#estimated group prior variance (all iterations)
estimated_group_prior_var_all <- group_prior_var_rec
#set group size
group_size <- c(table(ctwas_gene_res$type), structure(n_snps, names="SNP"))
group_size <- group_size[rownames(estimated_group_prior_all)]
#estimated group PVE (all iterations)
estimated_group_pve_all <- estimated_group_prior_var_all*estimated_group_prior_all*group_size/sample_size #check PVE calculation
results_current <- data.frame(simutag=as.character(simutag),
n_causal=as.integer(n_causal),
n_causal_combined=as.integer(n_causal_combined),
n_detected_pip=as.integer(n_ctwas_genes),
n_detected_pip_in_causal=as.integer(n_causal_detected),
n_detected_comb_pip=as.integer(n_ctwas_genes_combined),
n_detected_comb_pip_in_causal=as.integer(n_causal_detected_combined),
pve_snp=as.numeric(rev(estimated_group_pve_all["SNP",])[1]),
pve_weight1=as.numeric(rev(estimated_group_pve_all["Brain_Cerebellum_harmonized",])[1]),
pve_weight2=as.numeric(rev(estimated_group_pve_all["Brain_Hippocampus_harmonized",])[1]),
pve_weight3=as.numeric(rev(estimated_group_pve_all["Brain_Caudate_basal_ganglia_harmonized",])[1]),
prior_snp=as.numeric(rev(estimated_group_prior_var_all["SNP",])[1]),
prior_weight1=as.numeric(rev(estimated_group_prior_all["Brain_Cerebellum_harmonized",])[1]),
prior_weight2=as.numeric(rev(estimated_group_prior_all["Brain_Hippocampus_harmonized",])[1]),
prior_weight3=as.numeric(rev(estimated_group_prior_all["Brain_Caudate_basal_ganglia_harmonized",])[1]),
prior_var_snp=as.numeric(rev(estimated_group_prior_var_all["SNP",])[1]),
prior_var_weight1=as.numeric(rev(estimated_group_prior_var_all["Brain_Cerebellum_harmonized",])[1]),
prior_var_weight2=as.numeric(rev(estimated_group_prior_var_all["Brain_Hippocampus_harmonized",])[1]),
prior_var_weight3=as.numeric(rev(estimated_group_prior_var_all["Brain_Caudate_basal_ganglia_harmonized",])[1]))
results_df <- rbind(results_df, results_current)
}
#results using PIP threshold (gene+tissue)
results_df[,c("simutag", "n_causal", "n_detected_pip", "n_detected_pip_in_causal")] simutag n_causal n_detected_pip n_detected_pip_in_causal
1 4-1 90 34 13
2 4-2 105 47 11
3 4-3 114 64 25#mean percent causal using PIP > 0.8
sum(results_df$n_detected_pip_in_causal)/sum(results_df$n_detected_pip)[1] 0.337931#results using combined PIP threshold
results_df[,c("simutag", "n_causal_combined", "n_detected_comb_pip", "n_detected_comb_pip_in_causal")] simutag n_causal_combined n_detected_comb_pip n_detected_comb_pip_in_causal
1 4-1 90 37 15
2 4-2 105 55 20
3 4-3 114 67 26#mean percent causal using combined PIP > 0.8
sum(results_df$n_detected_comb_pip_in_causal)/sum(results_df$n_detected_comb_pip)[1] 0.3836478#prior inclusion and mean prior inclusion
results_df[,c(which(colnames(results_df)=="simutag"), setdiff(grep("prior", names(results_df)), grep("prior_var", names(results_df))))] simutag prior_snp prior_weight1 prior_weight2 prior_weight3
1 4-1 8.581569 0.006602027 0.0021024441 0.001099093
2 4-2 8.087527 0.008219075 0.0007509542 0.005859247
3 4-3 7.605934 0.013902915 0.0053234702 0.003794378colMeans(results_df[,setdiff(grep("prior", names(results_df)), grep("prior_var", names(results_df)))]) prior_snp prior_weight1 prior_weight2 prior_weight3
8.091677013 0.009574672 0.002725623 0.003584240 #prior variance and mean prior variance
results_df[,c(which(colnames(results_df)=="simutag"), grep("prior_var", names(results_df)))] simutag prior_var_snp prior_var_weight1 prior_var_weight2 prior_var_weight3
1 4-1 8.581569 16.43984 14.88672 1.800665
2 4-2 8.087527 15.93081 20.36791 19.809020
3 4-3 7.605934 15.17775 12.01800 3.115657colMeans(results_df[,grep("prior_var", names(results_df))]) prior_var_snp prior_var_weight1 prior_var_weight2 prior_var_weight3
8.091677 15.849465 15.757544 8.241781 #PVE and mean PVE
results_df[,c(which(colnames(results_df)=="simutag"), grep("pve", names(results_df)))] simutag pve_snp pve_weight1 pve_weight2 pve_weight3
1 4-1 0.2517062 0.02115492 0.005529403 0.0003773922
2 4-2 0.2586432 0.02552098 0.002702181 0.0221324851
3 4-3 0.2482345 0.04112916 0.011302685 0.0022543205colMeans(results_df[,grep("pve", names(results_df))]) pve_snp pve_weight1 pve_weight2 pve_weight3
0.252861290 0.029268354 0.006511423 0.008254733 #store results for figure
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS G - Ind Var",
count=results_df$n_detected_comb_pip-results_df$n_detected_comb_pip_in_causal,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS G - Ind Var",
count=results_df$n_detected_comb_pip_in_causal,
ifcausal=T))Individual tissue analyses
For the cTWAS analysis, each tissue was analyzed individually and the results were combined.
results_dir <- "/project2/xinhe/shengqian/cTWAS/cTWAS_simulation/simulation_correlated/"
runtag = "ukb-s80.45-3_corr"
configtag <- 2
simutags <- paste(4, 1:3, sep = "-")
thin <- 0.1
sample_size <- 45000
PIP_threshold <- 0.8
results_df <- data.frame(simutag=as.character(),
n_causal_combined=as.integer(),
n_detected_weight1=as.integer(),
n_detected_in_causal_weight1=as.integer(),
n_detected_weight2=as.integer(),
n_detected_in_causal_weight2=as.integer(),
n_detected_weight3=as.integer(),
n_detected_in_causal_weight3=as.integer(),
n_detected_combined=as.integer(),
n_detected_in_causal_combined=as.integer())
for (i in 1:length(simutags)){
simutag <- simutags[i]
#load genes with true simulated effect
load(paste0(results_dir, runtag, "_simu", simutag, "-pheno.Rd"))
true_genes <- unlist(sapply(1:22, function(x){phenores$batch[[x]]$id.cgene}))
true_genes_combined <- unique(sapply(true_genes, function(x){unlist(strsplit(x, "[|]"))[1]}))
#number of causal genes
n_causal_combined <- length(true_genes_combined)
#load cTWAS results for weight1
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight1.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight1
ctwas_genes_weight1 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight1 <- length(ctwas_genes_weight1)
n_causal_detected_weight1 <- sum(ctwas_genes_weight1 %in% true_genes_combined)
#load cTWAS results for weight2
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight2.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight1
ctwas_genes_weight2 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight2 <- length(ctwas_genes_weight2)
n_causal_detected_weight2 <- sum(ctwas_genes_weight2 %in% true_genes_combined)
#load cTWAS results for weight3
ctwas_res <- data.table::fread(paste0(results_dir, runtag, "_simu", simutag, "_config", configtag, "_LDR_weight3.susieIrss.txt"))
ctwas_gene_res <- ctwas_res[ctwas_res$type!="SNP",]
#number of genes with cTWAS PIP > threshold in weight3
ctwas_genes_weight3 <- ctwas_gene_res$id[ctwas_gene_res$susie_pip > PIP_threshold]
n_ctwas_genes_weight3 <- length(ctwas_genes_weight3)
n_causal_detected_weight3 <- sum(ctwas_genes_weight3 %in% true_genes_combined)
#combined analysis
ctwas_genes_combined <- unique(c(ctwas_genes_weight1, ctwas_genes_weight2, ctwas_genes_weight3))
n_ctwas_genes_combined <- length(ctwas_genes_combined)
n_causal_detected_combined <- sum(ctwas_genes_combined %in% true_genes_combined)
results_current <- data.frame(simutag=as.character(simutag),
n_causal_combined=as.integer(n_causal_combined),
n_detected_weight1=as.integer(n_ctwas_genes_weight1),
n_detected_in_causal_weight1=as.integer(n_causal_detected_weight1),
n_detected_weight2=as.integer(n_ctwas_genes_weight2),
n_detected_in_causal_weight2=as.integer(n_causal_detected_weight2),
n_detected_weight3=as.integer(n_ctwas_genes_weight3),
n_detected_in_causal_weight3=as.integer(n_causal_detected_weight3),
n_detected_combined=as.integer(n_ctwas_genes_combined),
n_detected_in_causal_combined=as.integer(n_causal_detected_combined))
results_df <- rbind(results_df, results_current)
}
#results using weight1
results_df[,c("simutag", colnames(results_df)[grep("weight1", colnames(results_df))])] simutag n_detected_weight1 n_detected_in_causal_weight1
1 4-1 25 14
2 4-2 36 11
3 4-3 48 23#mean percent causal using PIP > 0.8 for weight1
sum(results_df$n_detected_in_causal_weight1)/sum(results_df$n_detected_weight1)[1] 0.440367#results using weight2
results_df[,c("simutag", colnames(results_df)[grep("weight2", colnames(results_df))])] simutag n_detected_weight2 n_detected_in_causal_weight2
1 4-1 22 6
2 4-2 29 13
3 4-3 32 9#mean percent causal using PIP > 0.8 for weight1
sum(results_df$n_detected_in_causal_weight2)/sum(results_df$n_detected_weight2)[1] 0.3373494#results using weight3
results_df[,c("simutag", colnames(results_df)[grep("weight3", colnames(results_df))])] simutag n_detected_weight3 n_detected_in_causal_weight3
1 4-1 16 4
2 4-2 21 5
3 4-3 5 3#mean percent causal using PIP > 0.8 for weight3
sum(results_df$n_detected_in_causal_weight3)/sum(results_df$n_detected_weight3)[1] 0.2857143#results using combined analysis
results_df[,c("simutag", colnames(results_df)[grep("combined", colnames(results_df))])] simutag n_causal_combined n_detected_combined n_detected_in_causal_combined
1 4-1 90 46 16
2 4-2 105 66 20
3 4-3 114 72 28#mean percent causal using PIP > 0.8 for weight3
sum(results_df$n_detected_in_causal_combined)/sum(results_df$n_detected_combined)[1] 0.3478261#store results for figure
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight1",
count=results_df$n_detected_weight1-results_df$n_detected_in_causal_weight1,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight1",
count=results_df$n_detected_in_causal_weight1,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight2",
count=results_df$n_detected_weight2-results_df$n_detected_in_causal_weight2,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight2",
count=results_df$n_detected_in_causal_weight2,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight3",
count=results_df$n_detected_weight3-results_df$n_detected_in_causal_weight3,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Weight3",
count=results_df$n_detected_in_causal_weight3,
ifcausal=T))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Union",
count=results_df$n_detected_combined-results_df$n_detected_in_causal_combined,
ifcausal=F))
plot_df <- rbind(plot_df,
data.frame(simutag=results_df$simutag,
method="cTWAS Union",
count=results_df$n_detected_in_causal_combined,
ifcausal=T))Figure
For the cTWAS analysis, each tissue was analyzed individually and the results were combined.
plot_df$method <- factor(plot_df$method, levels=c("TWAS", "cTWAS G+T", "cTWAS G", "cTWAS G - Ind Var", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3"))
library(ggpubr)
colset = c("#ebebeb", "#fb8072")
ggbarplot(plot_df,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "none",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1))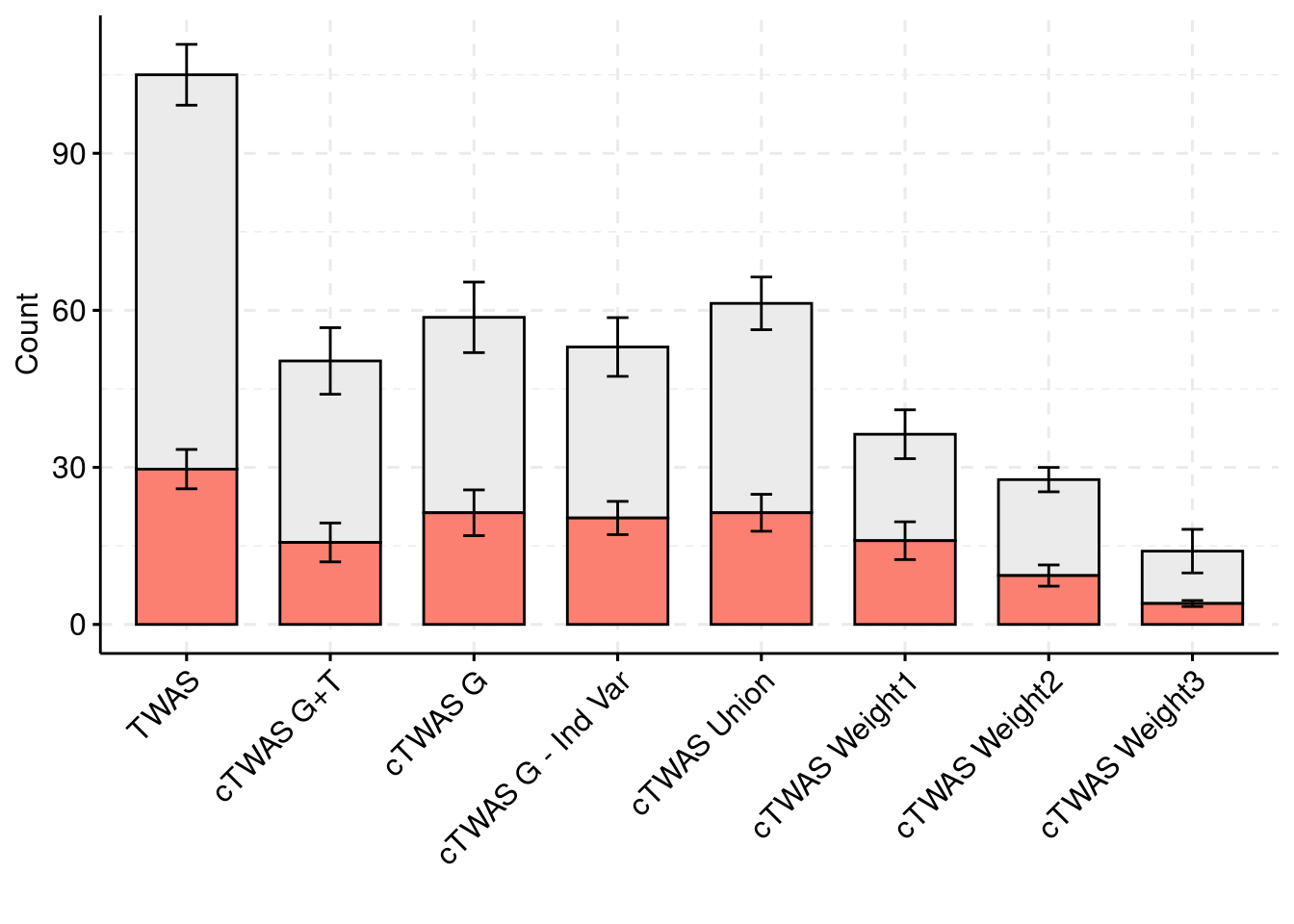
plot_df_2 <- plot_df
plot_df_2$method %in% c("TWAS", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3") [1] FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE
[13] TRUE TRUE TRUE TRUE TRUE TRUE FALSE FALSE FALSE FALSE FALSE FALSE
[25] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE
[37] TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUE TRUEplot_df_2$ifcausal <- as.character(plot_df_2$ifcausal+1)
plot_df_2$count[plot_df_2$method=="cTWAS G" & plot_df_2$ifcausal=="2"] <- plot_df_2$count[plot_df_2$method=="cTWAS G" & plot_df_2$ifcausal=="2"] - plot_df_2$count[plot_df_2$method=="cTWAS G+T" & plot_df_2$ifcausal=="2"]
plot_df_2 <- rbind(plot_df_2, data.frame(simutag=simutags,
method="cTWAS G",
count=plot_df_2$count[plot_df_2$method=="cTWAS G+T" & plot_df_2$ifcausal=="2"],
ifcausal="3"))
plot_df_3 <- plot_df_2
plot_df_2 <- plot_df_2[plot_df_2$method %in% c("TWAS", "cTWAS Union", "cTWAS Weight1", "cTWAS Weight2", "cTWAS Weight3"),]
plot_df_2$method <- droplevels(plot_df_2$method)
levels(plot_df_2$method) <- c("TWAS", "cTWAS Union", "cTWAS Primary", "cTWAS Secondary", "cTWAS Null")
plot_df_2$ifcausal[plot_df_2$ifcausal=="1"] <- "FP"
plot_df_2$ifcausal[plot_df_2$ifcausal=="2"] <- "TP"
colset = c("#ebebeb", "#fb8072", "#8dd3c7")
ggbarplot(plot_df_2,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "right",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1)) + guides(fill=guide_legend(title=""))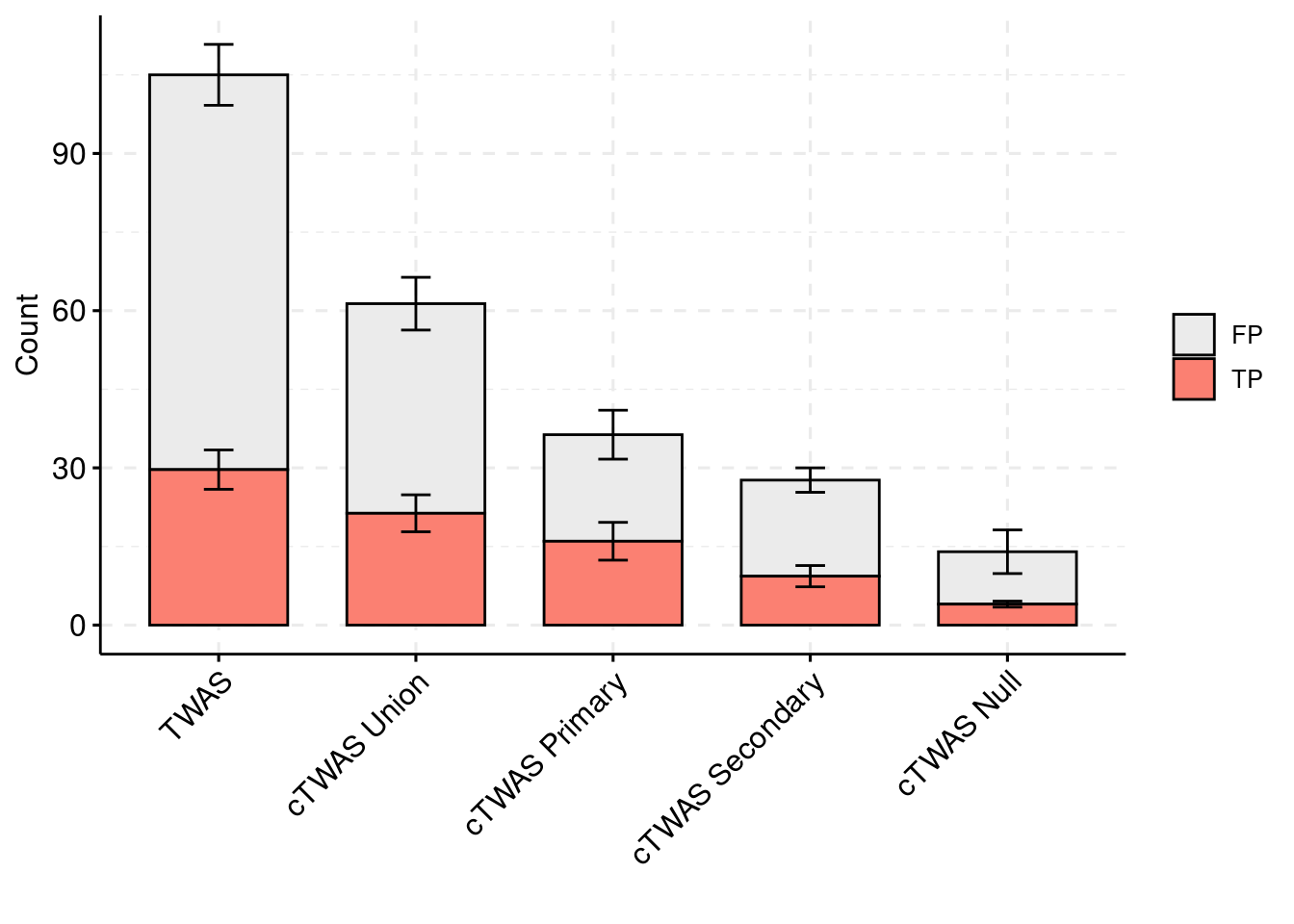
plot_df_3 <- plot_df_3[plot_df_3$method %in% c("cTWAS G", "cTWAS Union"),]
plot_df_3$method <- droplevels(plot_df_3$method)
levels(plot_df_3$method) <- c("cTWAS Joint", "cTWAS Union")
plot_df_3$method <- relevel(plot_df_3$method, "cTWAS Union")
plot_df_3$ifcausal[plot_df_3$ifcausal=="1"] <- "FP"
plot_df_3$ifcausal[plot_df_3$ifcausal=="2"] <- "TP"
plot_df_3$ifcausal[plot_df_3$ifcausal=="3"] <- "TP and Tissue Identified\nby Joint Analysis"
ggbarplot(plot_df_3,
x = "method",
y = "count",
add = "mean_se",
fill = "ifcausal",
legend = "right",
ylab="Count",
xlab="",
palette = colset) + grids(linetype = "dashed") + theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1)) + guides(fill=guide_legend(title=""))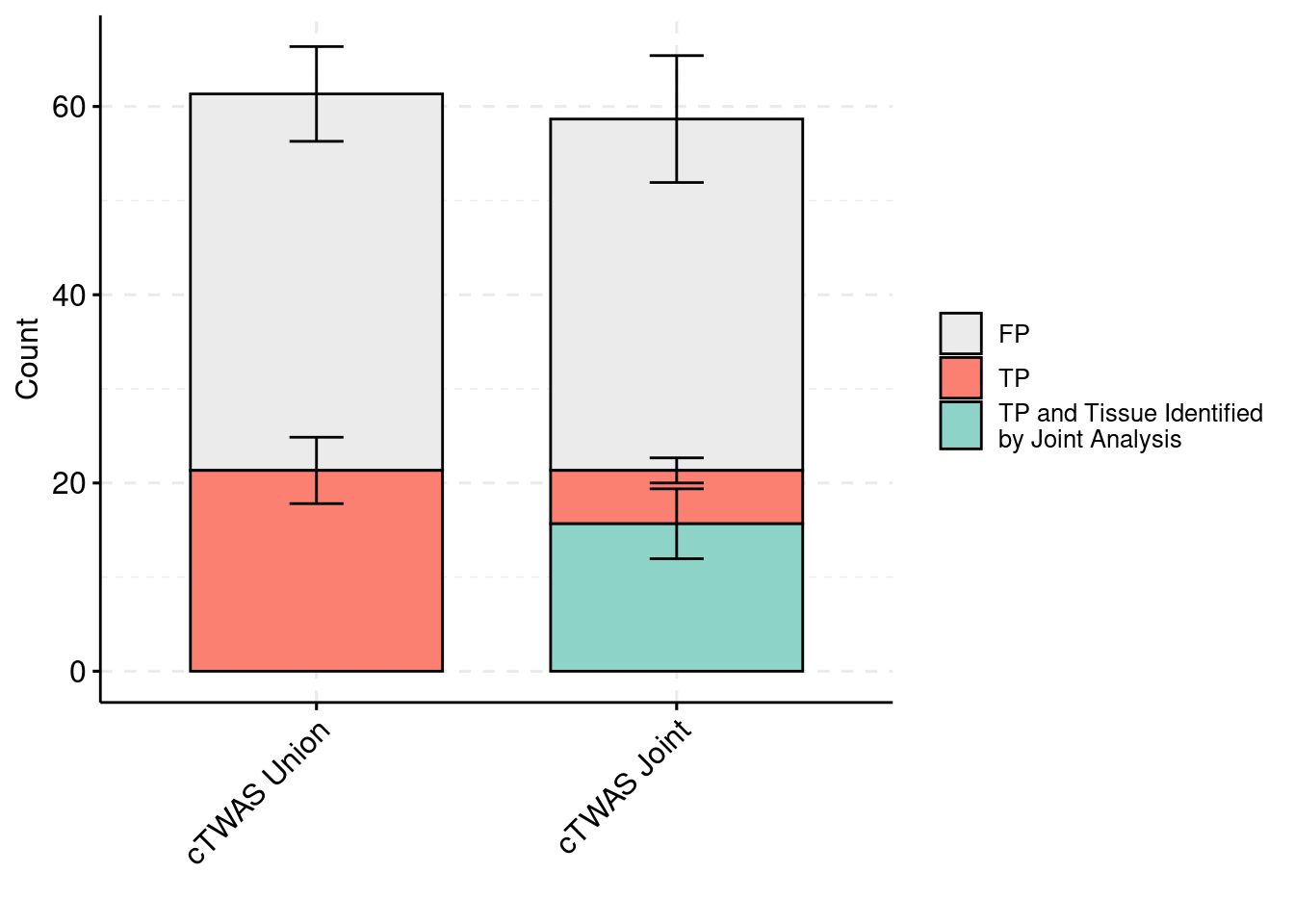
sessionInfo()R version 4.1.0 (2021-05-18)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggpubr_0.6.0 ggplot2_3.4.0 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] tidyselect_1.2.0 xfun_0.35 bslib_0.4.1 purrr_1.0.2
[5] carData_3.0-4 colorspace_2.0-3 vctrs_0.6.3 generics_0.1.3
[9] htmltools_0.5.4 yaml_2.3.6 utf8_1.2.2 rlang_1.1.1
[13] jquerylib_0.1.4 later_1.3.0 pillar_1.8.1 glue_1.6.2
[17] withr_2.5.0 DBI_1.1.3 lifecycle_1.0.3 stringr_1.5.0
[21] ggsignif_0.6.4 munsell_0.5.0 gtable_0.3.1 evaluate_0.19
[25] labeling_0.4.2 knitr_1.41 callr_3.7.3 fastmap_1.1.0
[29] httpuv_1.6.7 ps_1.7.2 fansi_1.0.3 highr_0.9
[33] broom_1.0.2 Rcpp_1.0.9 backports_1.2.1 promises_1.2.0.1
[37] scales_1.2.1 cachem_1.0.6 jsonlite_1.8.4 abind_1.4-5
[41] farver_2.1.0 fs_1.5.2 digest_0.6.31 stringi_1.7.8
[45] rstatix_0.7.2 processx_3.8.0 dplyr_1.0.10 getPass_0.2-2
[49] rprojroot_2.0.3 grid_4.1.0 cli_3.6.1 tools_4.1.0
[53] magrittr_2.0.3 sass_0.4.4 tibble_3.1.8 car_3.1-1
[57] tidyr_1.3.0 whisker_0.4.1 pkgconfig_2.0.3 data.table_1.14.6
[61] assertthat_0.2.1 rmarkdown_2.19 httr_1.4.4 rstudioapi_0.14
[65] R6_2.5.1 git2r_0.30.1 compiler_4.1.0