SCZ - Brain Anterior cingulate cortex BA24
sheng Qian
2021-2-6
Last updated: 2022-04-19
Checks: 5 2
Knit directory: cTWAS_analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20211220) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /project2/xinhe/shengqian/cTWAS/cTWAS_analysis/data/ | data |
| /project2/xinhe/shengqian/cTWAS/cTWAS_analysis/code/ctwas_config.R | code/ctwas_config.R |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version ba919ab. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .ipynb_checkpoints/
Ignored: data/AF/
Untracked files:
Untracked: Rplot.png
Untracked: analysis/.ipynb_checkpoints/
Untracked: code/.ipynb_checkpoints/
Untracked: code/AF_out/
Untracked: code/Autism_out/
Untracked: code/BMI_S_out/
Untracked: code/BMI_out/
Untracked: code/Glucose_out/
Untracked: code/LDL_S_out/
Untracked: code/SCZ_2014_EUR_out/
Untracked: code/SCZ_2018_out/
Untracked: code/SCZ_2020_Single_out/
Untracked: code/SCZ_2020_out/
Untracked: code/SCZ_S_out/
Untracked: code/SCZ_out/
Untracked: code/T2D_out/
Untracked: code/ctwas_config.R
Untracked: code/mapping.R
Untracked: code/out/
Untracked: code/process_scz_2018_snps.R
Untracked: code/run_AF_analysis.sbatch
Untracked: code/run_AF_analysis.sh
Untracked: code/run_AF_ctwas_rss_LDR.R
Untracked: code/run_Autism_analysis.sbatch
Untracked: code/run_Autism_analysis.sh
Untracked: code/run_Autism_ctwas_rss_LDR.R
Untracked: code/run_BMI_analysis.sbatch
Untracked: code/run_BMI_analysis.sh
Untracked: code/run_BMI_analysis_S.sbatch
Untracked: code/run_BMI_analysis_S.sh
Untracked: code/run_BMI_ctwas_rss_LDR.R
Untracked: code/run_BMI_ctwas_rss_LDR_S.R
Untracked: code/run_Glucose_analysis.sbatch
Untracked: code/run_Glucose_analysis.sh
Untracked: code/run_Glucose_ctwas_rss_LDR.R
Untracked: code/run_LDL_analysis_S.sbatch
Untracked: code/run_LDL_analysis_S.sh
Untracked: code/run_LDL_ctwas_rss_LDR_S.R
Untracked: code/run_SCZ_2014_EUR_analysis.sbatch
Untracked: code/run_SCZ_2014_EUR_analysis.sh
Untracked: code/run_SCZ_2014_EUR_ctwas_rss_LDR.R
Untracked: code/run_SCZ_2018_analysis.sbatch
Untracked: code/run_SCZ_2018_analysis.sh
Untracked: code/run_SCZ_2018_ctwas_rss_LDR.R
Untracked: code/run_SCZ_2020_Single_analysis.sbatch
Untracked: code/run_SCZ_2020_Single_analysis.sh
Untracked: code/run_SCZ_2020_Single_ctwas_rss_LDR.R
Untracked: code/run_SCZ_2020_analysis.sbatch
Untracked: code/run_SCZ_2020_analysis.sh
Untracked: code/run_SCZ_2020_ctwas_rss_LDR.R
Untracked: code/run_SCZ_analysis.sbatch
Untracked: code/run_SCZ_analysis.sh
Untracked: code/run_SCZ_analysis_S.sbatch
Untracked: code/run_SCZ_analysis_S.sh
Untracked: code/run_SCZ_ctwas_rss_LDR.R
Untracked: code/run_SCZ_ctwas_rss_LDR_S.R
Untracked: code/run_T2D_analysis.sbatch
Untracked: code/run_T2D_analysis.sh
Untracked: code/run_T2D_ctwas_rss_LDR.R
Untracked: code/wflow_build.R
Untracked: code/wflow_build.sbatch
Untracked: data/.ipynb_checkpoints/
Untracked: data/BMI/
Untracked: data/GO_Terms/
Untracked: data/PGC3_SCZ_wave3_public.v2.tsv
Untracked: data/SCZ/
Untracked: data/SCZ_2014_EUR/
Untracked: data/SCZ_2018/
Untracked: data/SCZ_2020/
Untracked: data/SCZ_2020_Single/
Untracked: data/SCZ_S/
Untracked: data/Supplementary Table 15 - MAGMA.xlsx
Untracked: data/Supplementary Table 20 - Prioritised Genes.xlsx
Untracked: data/T2D/
Untracked: data/UKBB/
Untracked: data/UKBB_SNPs_Info.text
Untracked: data/gene_OMIM.txt
Untracked: data/gene_pip_0.8.txt
Untracked: data/mashr_Heart_Atrial_Appendage.db
Untracked: data/mashr_sqtl/
Untracked: data/scz_2018.RDS
Untracked: data/summary_known_genes_annotations.xlsx
Untracked: data/untitled.txt
Untracked: top_genes_32.txt
Untracked: top_genes_37.txt
Untracked: top_genes_43.txt
Untracked: top_genes_81.txt
Unstaged changes:
Modified: analysis/SCZ_2018_Brain_Amygdala.Rmd
Modified: analysis/SCZ_2018_Brain_Anterior_cingulate_cortex_BA24.Rmd
Modified: analysis/SCZ_2018_Brain_Caudate_basal_ganglia.Rmd
Modified: analysis/SCZ_2018_Brain_Cerebellar_Hemisphere.Rmd
Modified: analysis/SCZ_2018_Brain_Cerebellum.Rmd
Modified: analysis/SCZ_2018_Brain_Cortex.Rmd
Modified: analysis/SCZ_2018_Brain_Frontal_Cortex_BA9.Rmd
Modified: analysis/SCZ_2018_Brain_Hippocampus.Rmd
Modified: analysis/SCZ_2018_Brain_Hypothalamus.Rmd
Modified: analysis/SCZ_2018_Brain_Nucleus_accumbens_basal_ganglia.Rmd
Modified: analysis/SCZ_2018_Brain_Putamen_basal_ganglia.Rmd
Modified: analysis/SCZ_2018_Brain_Spinal_cord_cervical_c-1.Rmd
Modified: analysis/SCZ_2018_Brain_Substantia_nigra.Rmd
Modified: analysis/SCZ_Annotation_Analysis.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/SCZ_2018_Brain_Anterior_cingulate_cortex_BA24.Rmd) and HTML (docs/SCZ_2018_Brain_Anterior_cingulate_cortex_BA24.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 9ddc9c4 | sq-96 | 2022-04-18 | update |
| Rmd | f6e7062 | sq-96 | 2022-04-17 | update |
| html | f6e7062 | sq-96 | 2022-04-17 | update |
Weight QC
#number of imputed weights
nrow(qclist_all)[1] 9453#number of imputed weights by chromosome
table(qclist_all$chr)
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
931 670 567 367 441 542 454 359 342 370 583 552 199 327 337 397 555 155 740 289
21 22
29 247 #number of imputed weights without missing variants
sum(qclist_all$nmiss==0)[1] 6793#proportion of imputed weights without missing variants
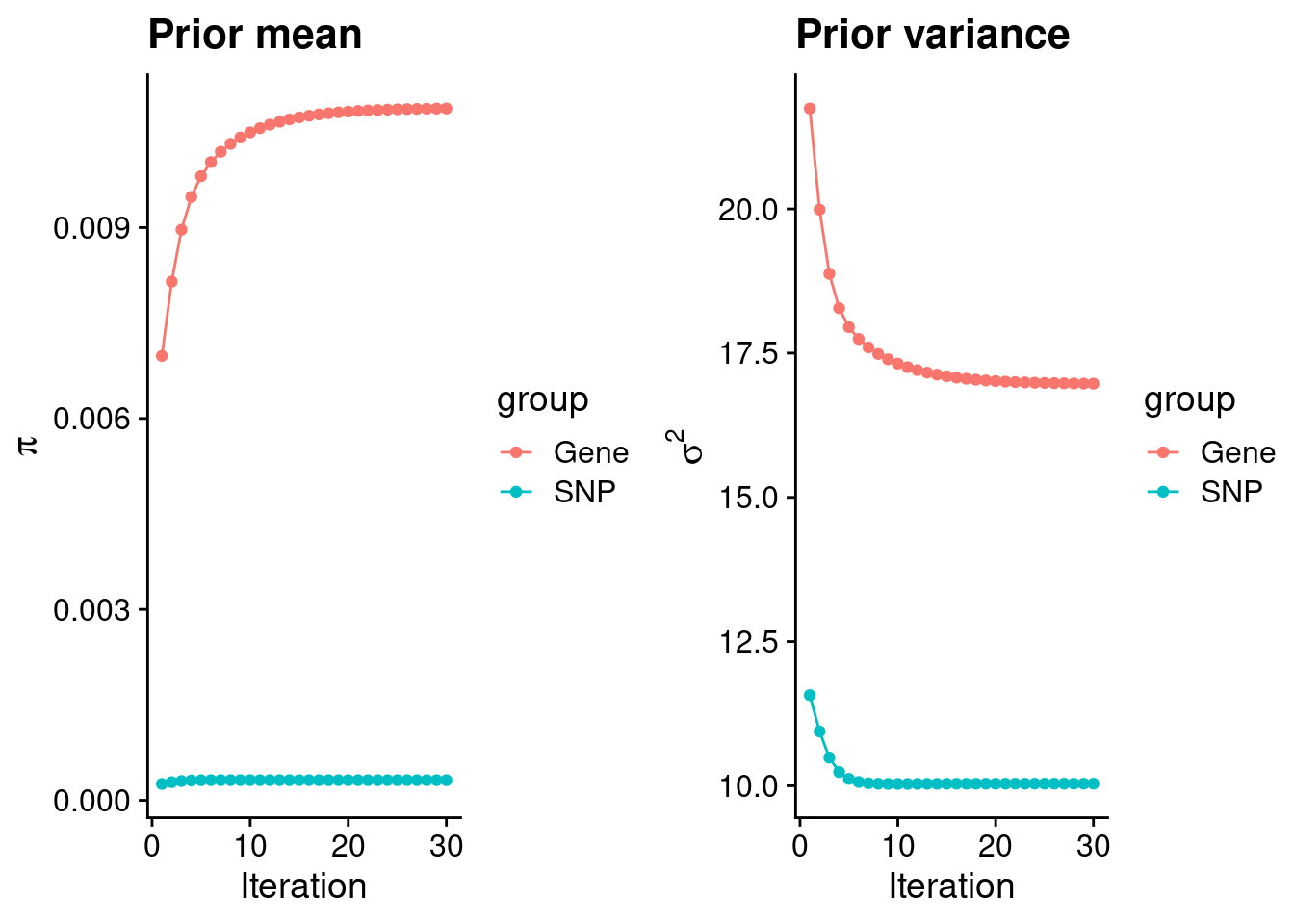
mean(qclist_all$nmiss==0)[1] 0.7186Check convergence of parameters
#estimated group prior
estimated_group_prior <- group_prior_rec[,ncol(group_prior_rec)]
names(estimated_group_prior) <- c("gene", "snp")
estimated_group_prior["snp"] <- estimated_group_prior["snp"]*thin #adjust parameter to account for thin argument
print(estimated_group_prior) gene snp
0.0108717 0.0003169 #estimated group prior variance
estimated_group_prior_var <- group_prior_var_rec[,ncol(group_prior_var_rec)]
names(estimated_group_prior_var) <- c("gene", "snp")
print(estimated_group_prior_var) gene snp
16.97 10.04 #report sample size
print(sample_size)[1] 105318#report group size
group_size <- c(nrow(ctwas_gene_res), n_snps)
print(group_size)[1] 9453 6309950#estimated group PVE
estimated_group_pve <- estimated_group_prior_var*estimated_group_prior*group_size/sample_size #check PVE calculation
names(estimated_group_pve) <- c("gene", "snp")
print(estimated_group_pve) gene snp
0.01656 0.19057 #compare sum(PIP*mu2/sample_size) with above PVE calculation
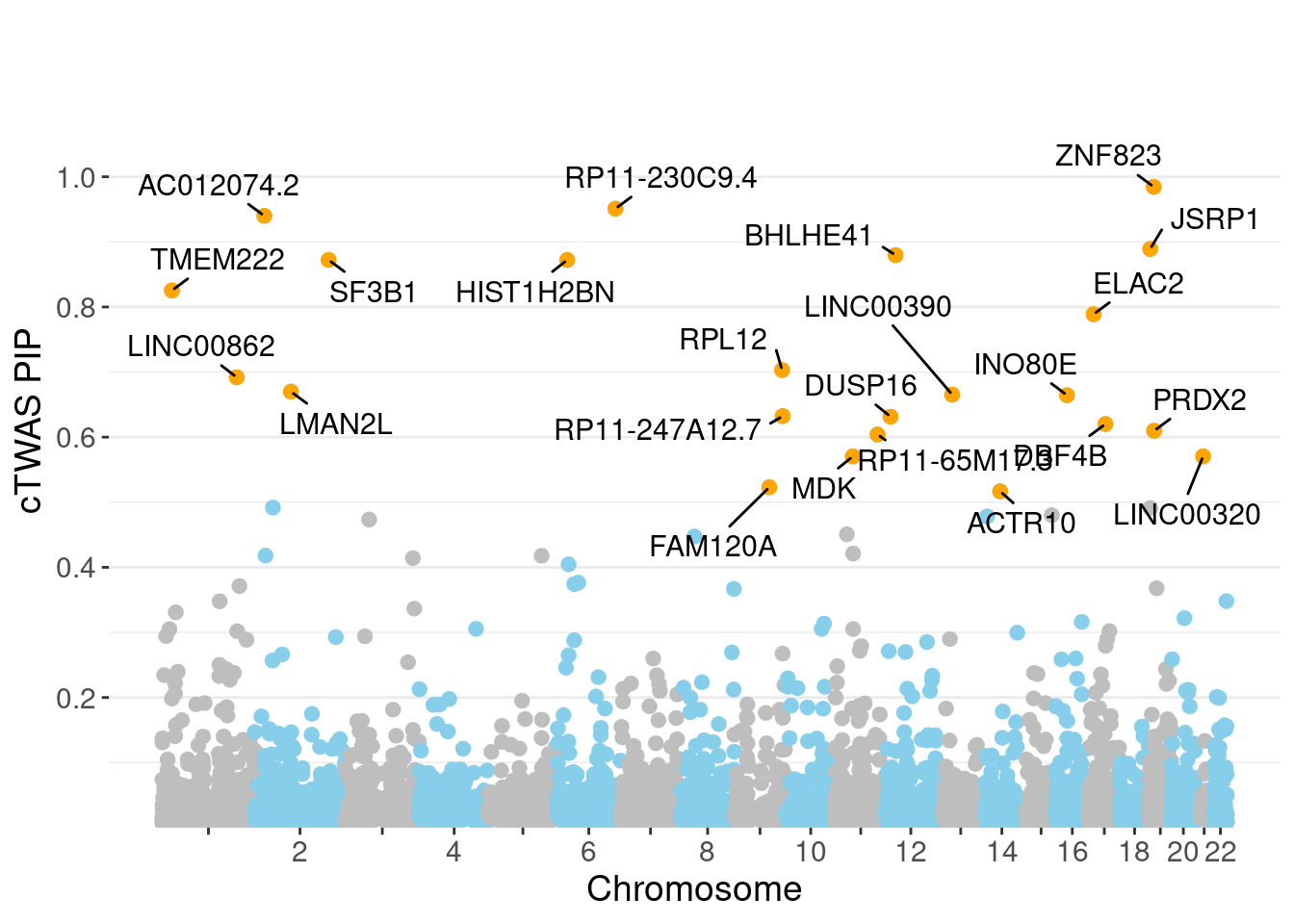
c(sum(ctwas_gene_res$PVE),sum(ctwas_snp_res$PVE))[1] 0.04975 1.07745Genes with highest PIPs
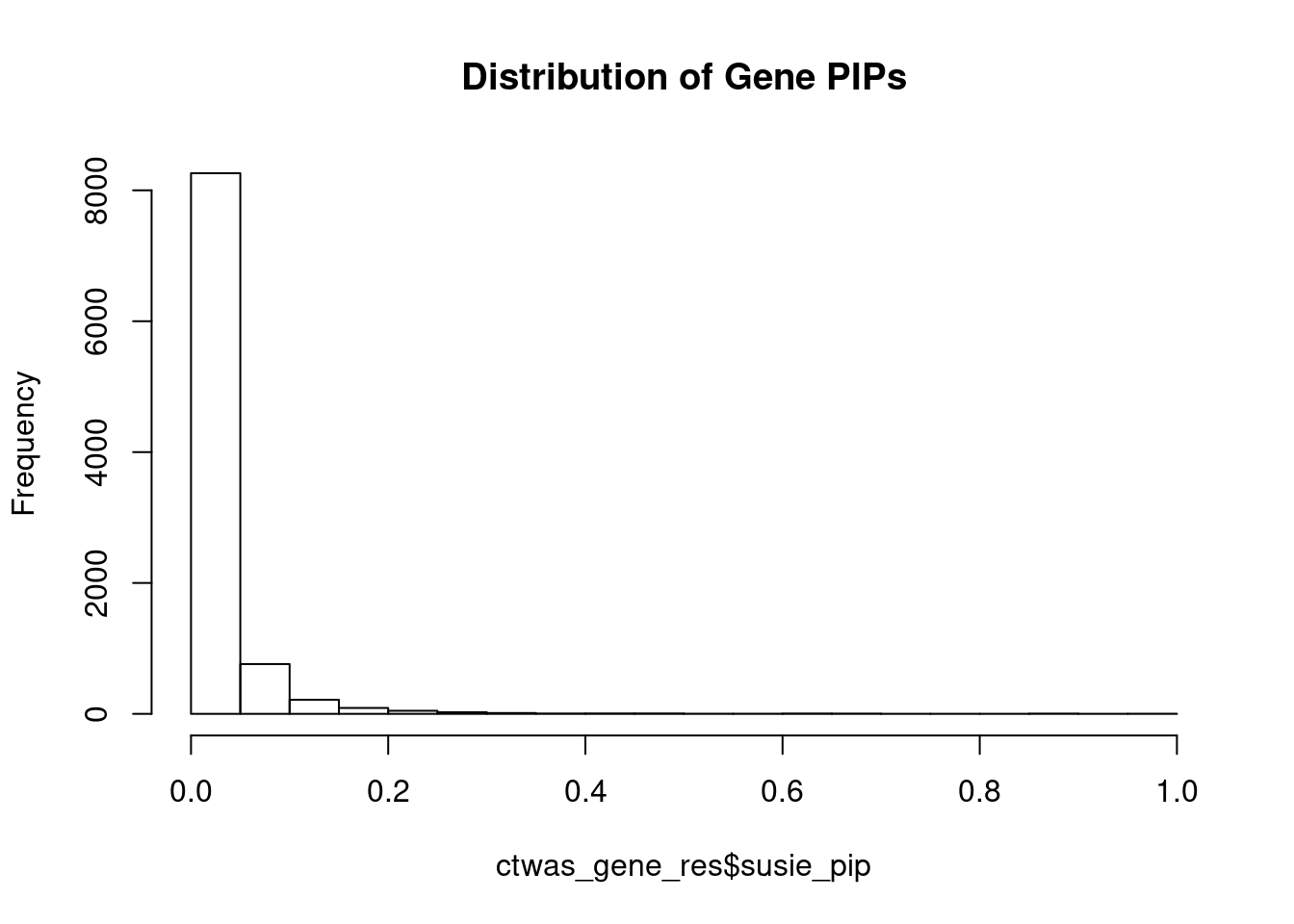
genename region_tag susie_pip mu2 PVE z num_eqtl
10719 ZNF823 19_10 0.9845 38.35 0.0003585 6.180 2
12952 RP11-230C9.4 6_102 0.9510 23.70 0.0002140 -4.685 2
11817 AC012074.2 2_15 0.9399 23.13 0.0002064 4.655 1
7971 JSRP1 19_3 0.8892 27.65 0.0002335 4.825 1
3590 BHLHE41 12_18 0.8795 24.00 0.0002004 4.516 1
2969 SF3B1 2_117 0.8722 51.50 0.0004265 7.265 1
11773 HIST1H2BN 6_21 0.8722 108.20 0.0008960 13.396 1
10112 TMEM222 1_19 0.8254 22.21 0.0001740 4.303 1
108 ELAC2 17_11 0.7888 22.90 0.0001715 4.752 1
10725 RPL12 9_66 0.7031 22.54 0.0001505 4.070 2
10939 LINC00862 1_101 0.6920 24.02 0.0001578 4.314 2
2898 LMAN2L 2_57 0.6699 25.27 0.0001607 -4.313 2
11543 LINC00390 13_17 0.6653 23.17 0.0001464 -4.540 1
8291 INO80E 16_24 0.6644 48.98 0.0003090 6.995 1
12860 RP11-247A12.7 9_66 0.6323 23.31 0.0001400 4.468 2
2584 DUSP16 12_11 0.6315 21.72 0.0001302 -3.779 1
7048 DBF4B 17_26 0.6202 20.50 0.0001207 3.890 1
8043 PRDX2 19_10 0.6098 22.76 0.0001318 -4.020 1
12334 RP11-65M17.3 11_66 0.6040 22.85 0.0001311 4.414 1
2524 MDK 11_28 0.5704 49.01 0.0002654 -7.159 1Genes with largest effect sizes
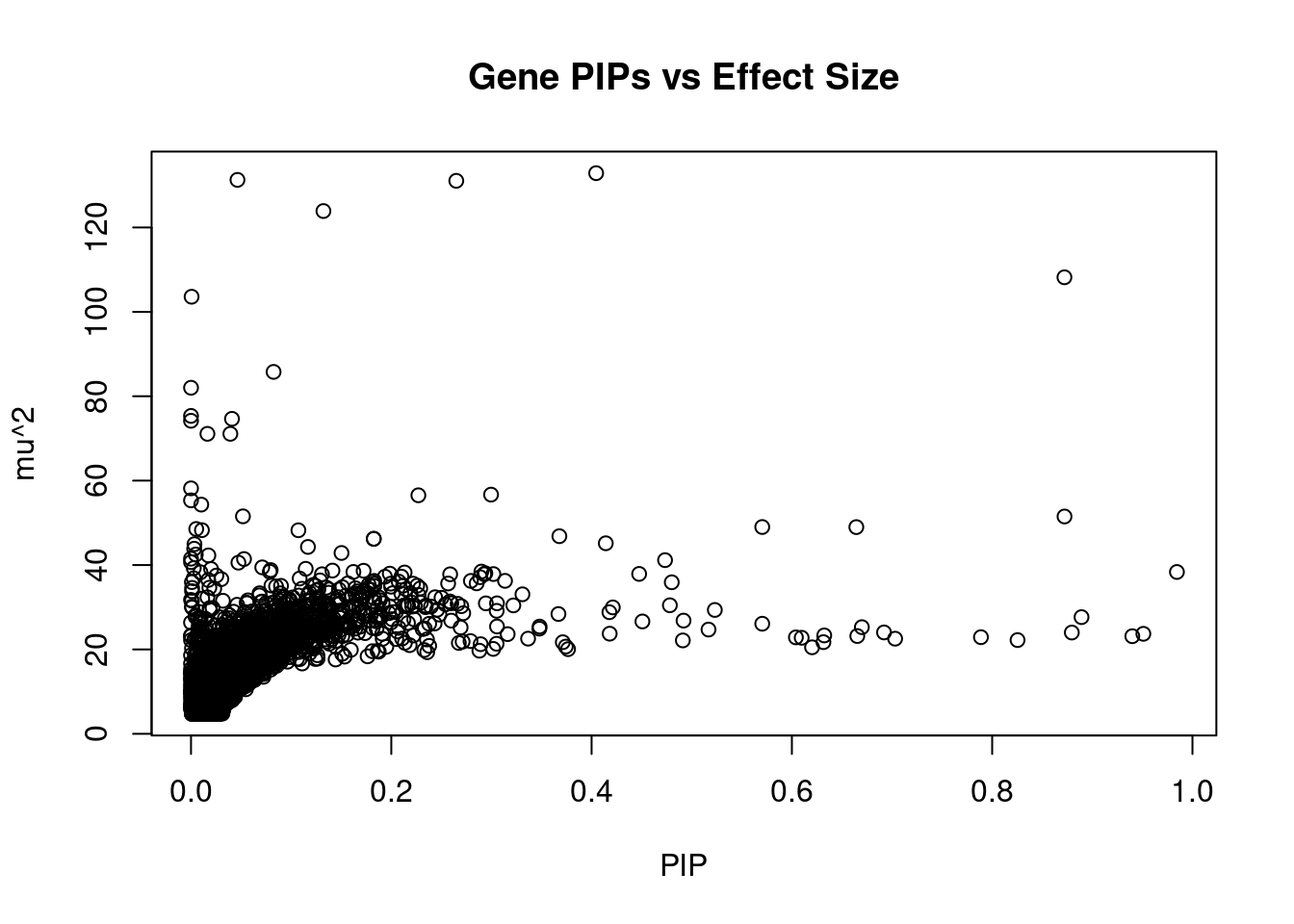
genename region_tag susie_pip mu2 PVE z num_eqtl
11039 APOM 6_26 4.046e-01 132.87 5.104e-04 11.590 1
12066 C4A 6_26 4.639e-02 131.28 5.783e-05 11.341 2
11035 ABHD16A 6_26 2.649e-01 131.06 3.296e-04 11.526 1
4944 VARS2 6_25 1.322e-01 123.89 1.555e-04 11.413 1
11773 HIST1H2BN 6_21 8.722e-01 108.20 8.960e-04 13.396 1
11008 NOTCH4 6_26 5.774e-04 103.58 5.679e-07 7.712 2
4935 FLOT1 6_24 8.245e-02 85.77 6.715e-05 -10.981 1
11034 LY6G6C 6_26 1.682e-05 82.01 1.310e-08 9.781 2
10534 HLA-DQA1 6_26 4.276e-06 75.34 3.059e-09 3.389 1
13060 RP1-86C11.7 6_21 4.090e-02 74.64 2.899e-05 10.889 1
9388 HLA-DQB1 6_26 7.594e-08 74.18 5.349e-11 4.986 1
11000 HLA-DMA 6_27 3.928e-02 71.09 2.652e-05 -8.720 2
10109 BTN3A2 6_20 1.638e-02 71.09 1.106e-05 9.166 2
10416 HLA-DRB1 6_26 8.466e-09 58.17 4.676e-12 1.359 2
1184 PPP1R13B 14_54 2.996e-01 56.68 1.612e-04 -6.610 2
455 MPHOSPH9 12_75 2.270e-01 56.53 1.219e-04 7.662 1
11277 DDAH2 6_26 1.744e-07 55.33 9.159e-11 8.149 1
10785 ZKSCAN8 6_22 1.025e-02 54.32 5.285e-06 7.317 2
10253 ZSCAN23 6_22 5.184e-02 51.54 2.537e-05 -7.854 1
2969 SF3B1 2_117 8.722e-01 51.50 4.265e-04 7.265 1Genes with highest PVE
genename region_tag susie_pip mu2 PVE z num_eqtl
11773 HIST1H2BN 6_21 0.8722 108.20 0.0008960 13.396 1
11039 APOM 6_26 0.4046 132.87 0.0005104 11.590 1
2969 SF3B1 2_117 0.8722 51.50 0.0004265 7.265 1
10719 ZNF823 19_10 0.9845 38.35 0.0003585 6.180 2
11035 ABHD16A 6_26 0.2649 131.06 0.0003296 11.526 1
8291 INO80E 16_24 0.6644 48.98 0.0003090 6.995 1
2524 MDK 11_28 0.5704 49.01 0.0002654 -7.159 1
7971 JSRP1 19_3 0.8892 27.65 0.0002335 4.825 1
12952 RP11-230C9.4 6_102 0.9510 23.70 0.0002140 -4.685 2
11817 AC012074.2 2_15 0.9399 23.13 0.0002064 4.655 1
3590 BHLHE41 12_18 0.8795 24.00 0.0002004 4.516 1
7325 THOC7 3_43 0.4734 41.15 0.0001850 -6.249 1
5786 CCDC39 3_111 0.4142 45.15 0.0001776 -6.797 1
10112 TMEM222 1_19 0.8254 22.21 0.0001740 4.303 1
108 ELAC2 17_11 0.7888 22.90 0.0001715 4.752 1
5316 FURIN 15_42 0.4801 35.91 0.0001637 -5.772 1
7973 GATAD2A 19_15 0.3678 46.83 0.0001636 -6.577 2
1184 PPP1R13B 14_54 0.2996 56.68 0.0001612 -6.610 2
13014 RP11-350N15.5 8_34 0.4475 37.89 0.0001610 5.963 1
2898 LMAN2L 2_57 0.6699 25.27 0.0001607 -4.313 2Genes with largest z scores
genename region_tag susie_pip mu2 PVE z num_eqtl
11773 HIST1H2BN 6_21 8.722e-01 108.20 8.960e-04 13.396 1
11039 APOM 6_26 4.046e-01 132.87 5.104e-04 11.590 1
11035 ABHD16A 6_26 2.649e-01 131.06 3.296e-04 11.526 1
4944 VARS2 6_25 1.322e-01 123.89 1.555e-04 11.413 1
12066 C4A 6_26 4.639e-02 131.28 5.783e-05 11.341 2
4935 FLOT1 6_24 8.245e-02 85.77 6.715e-05 -10.981 1
13060 RP1-86C11.7 6_21 4.090e-02 74.64 2.899e-05 10.889 1
11034 LY6G6C 6_26 1.682e-05 82.01 1.310e-08 9.781 2
10109 BTN3A2 6_20 1.638e-02 71.09 1.106e-05 9.166 2
11000 HLA-DMA 6_27 3.928e-02 71.09 2.652e-05 -8.720 2
6075 CNNM2 10_66 1.072e-01 48.24 4.911e-05 -8.161 1
11277 DDAH2 6_26 1.744e-07 55.33 9.159e-11 8.149 1
10253 ZSCAN23 6_22 5.184e-02 51.54 2.537e-05 -7.854 1
11008 NOTCH4 6_26 5.774e-04 103.58 5.679e-07 7.712 2
455 MPHOSPH9 12_75 2.270e-01 56.53 1.219e-04 7.662 1
10785 ZKSCAN8 6_22 1.025e-02 54.32 5.285e-06 7.317 2
2969 SF3B1 2_117 8.722e-01 51.50 4.265e-04 7.265 1
11051 POU5F1 6_25 1.734e-02 42.28 6.962e-06 -7.217 2
2524 MDK 11_28 5.704e-01 49.01 2.654e-04 -7.159 1
10542 ZSCAN16 6_22 1.090e-02 48.26 4.994e-06 7.135 1Comparing z scores and PIPs
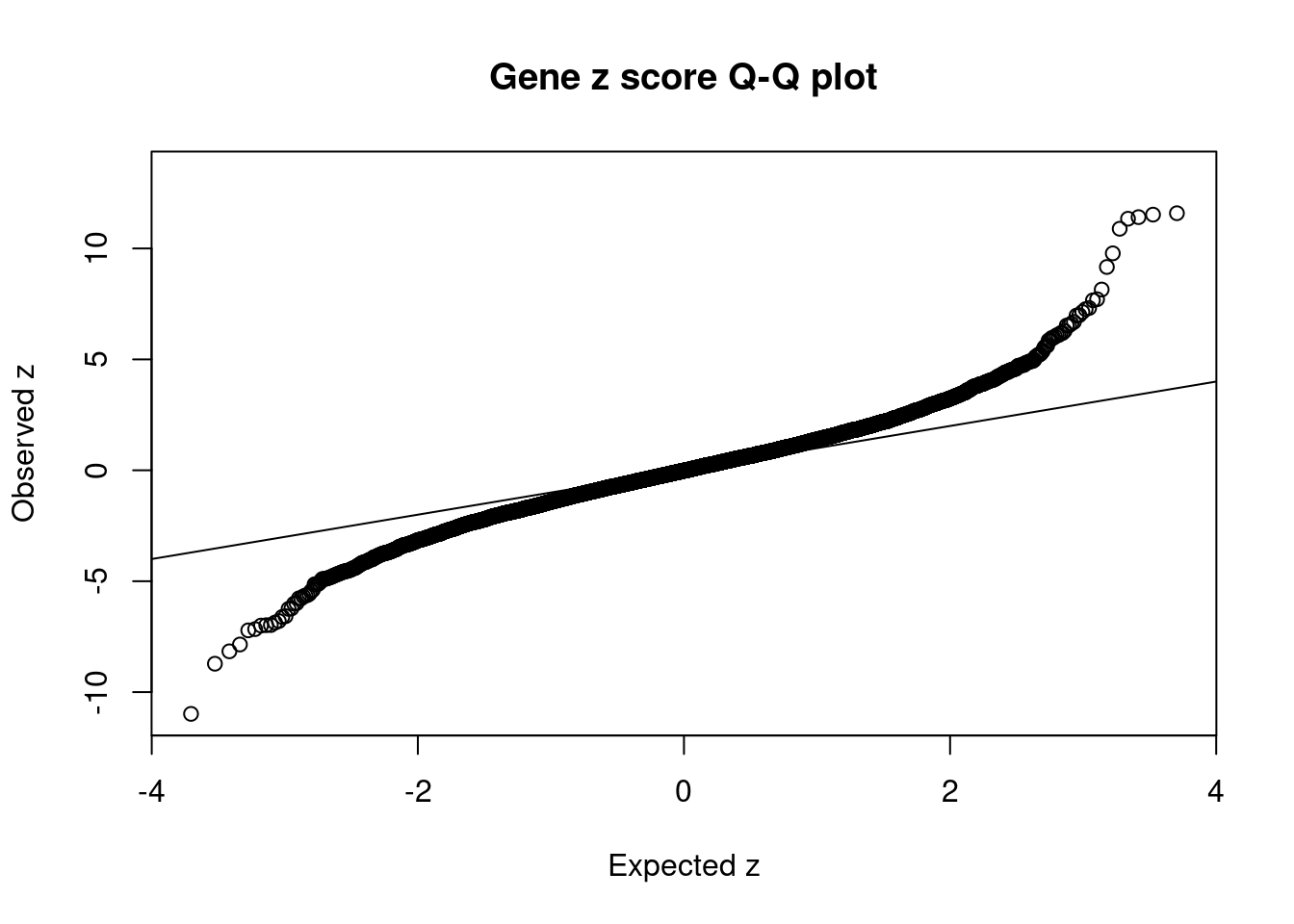
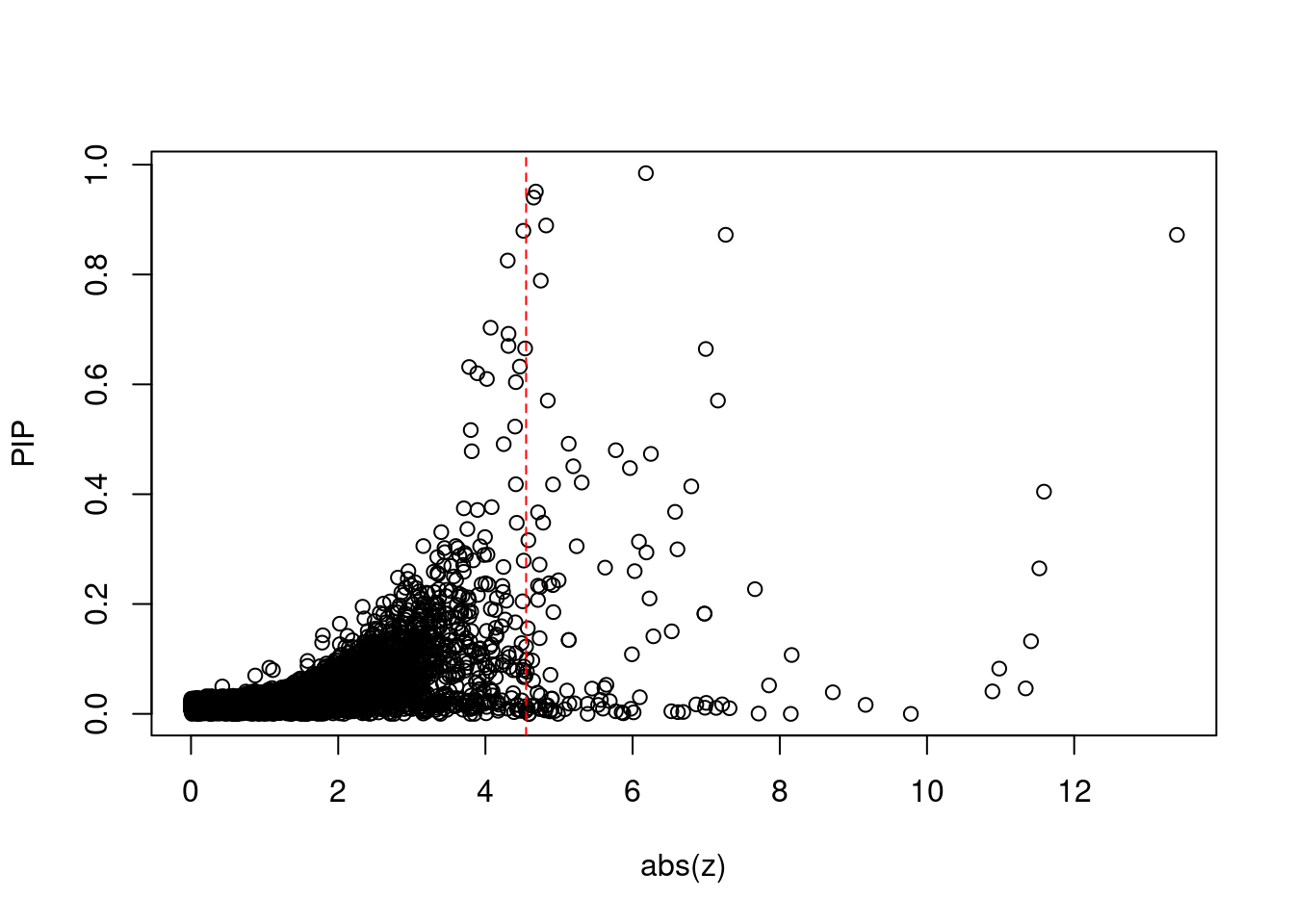
[1] 0.01195GO enrichment analysis for genes with PIP>0.5
#number of genes for gene set enrichment
length(genes)[1] 23Uploading data to Enrichr... Done.
Querying GO_Biological_Process_2021... Done.
Querying GO_Cellular_Component_2021... Done.
Querying GO_Molecular_Function_2021... Done.
Parsing results... Done.
[1] "GO_Biological_Process_2021"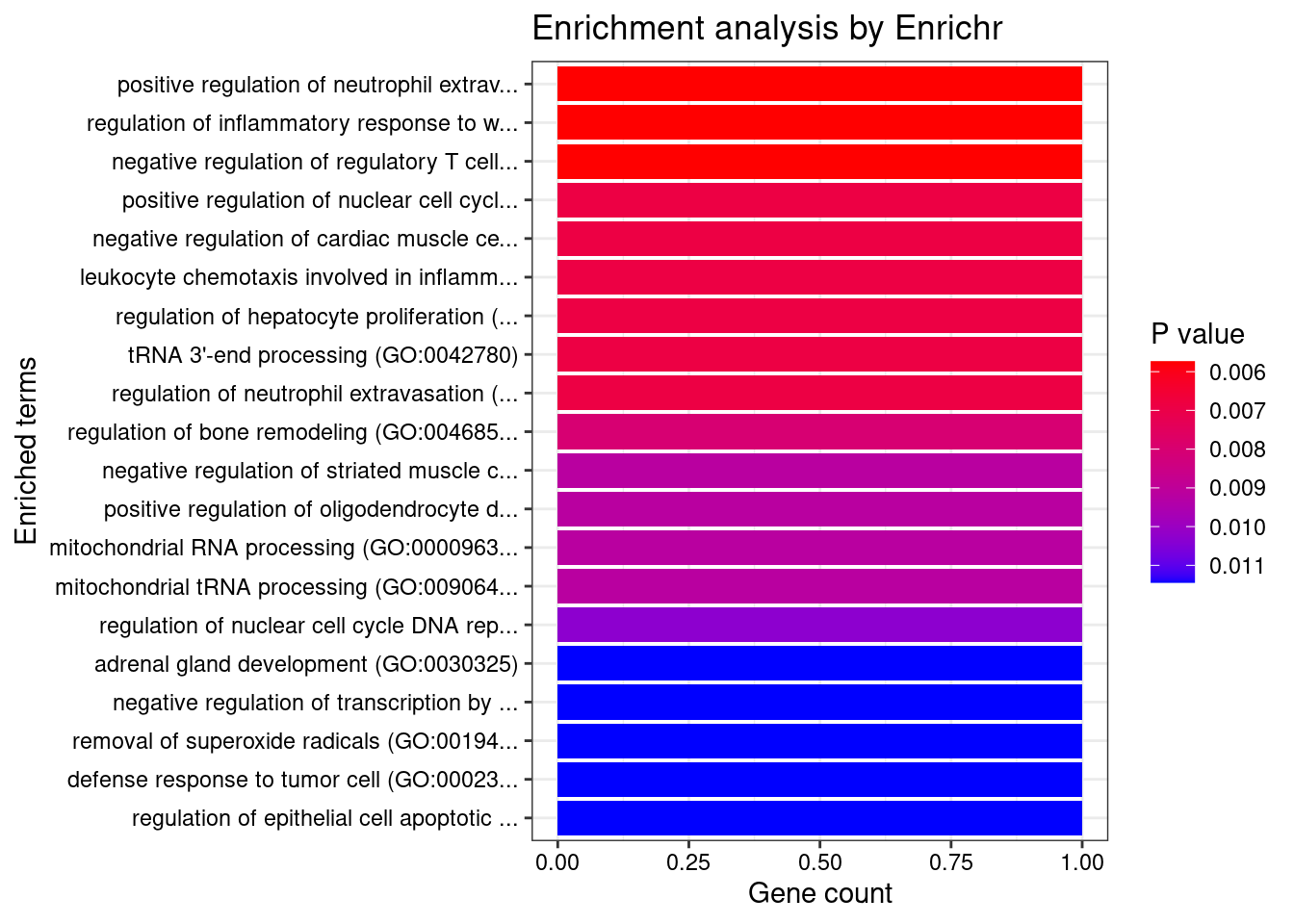
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Cellular_Component_2021"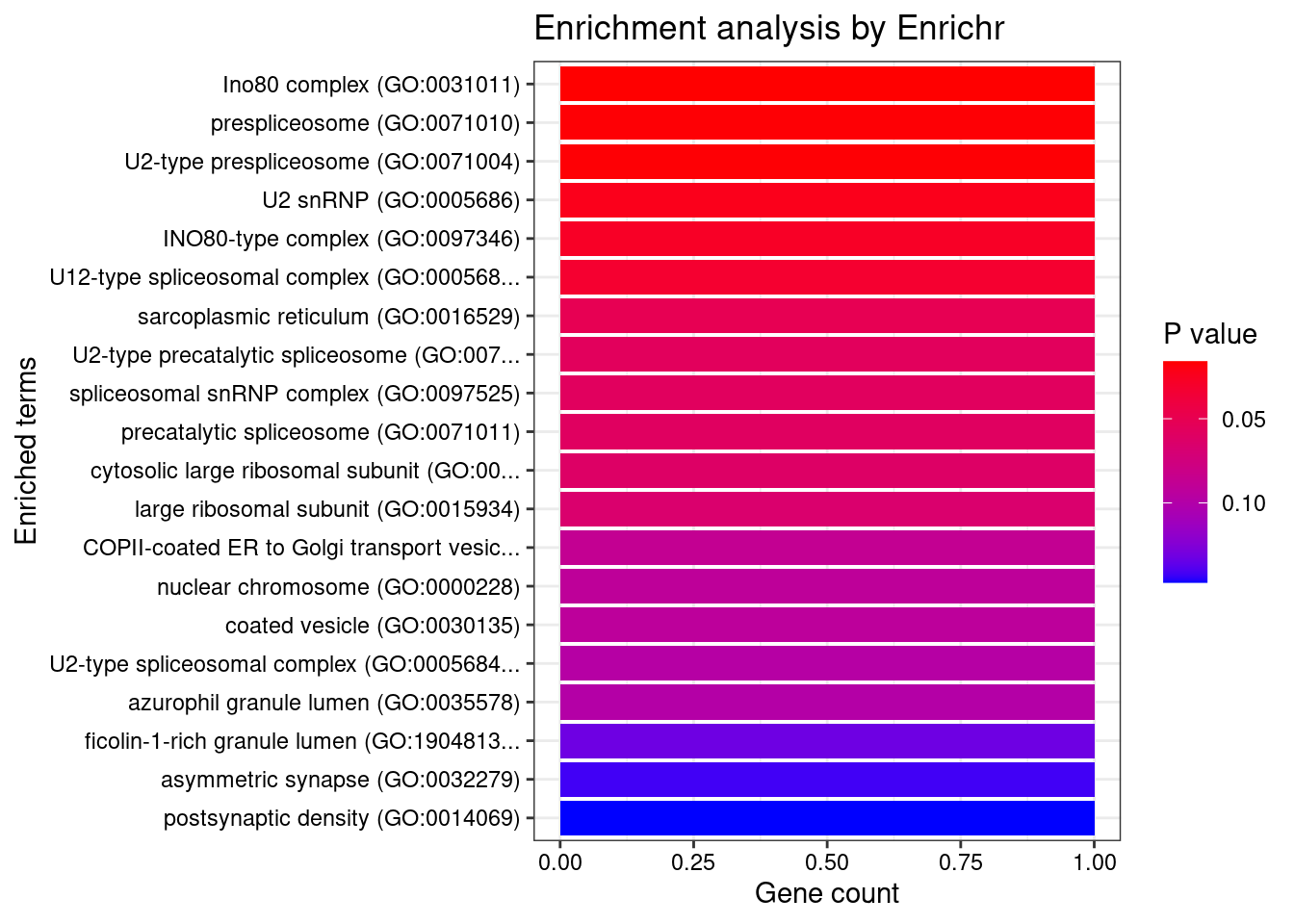
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)
[1] "GO_Molecular_Function_2021"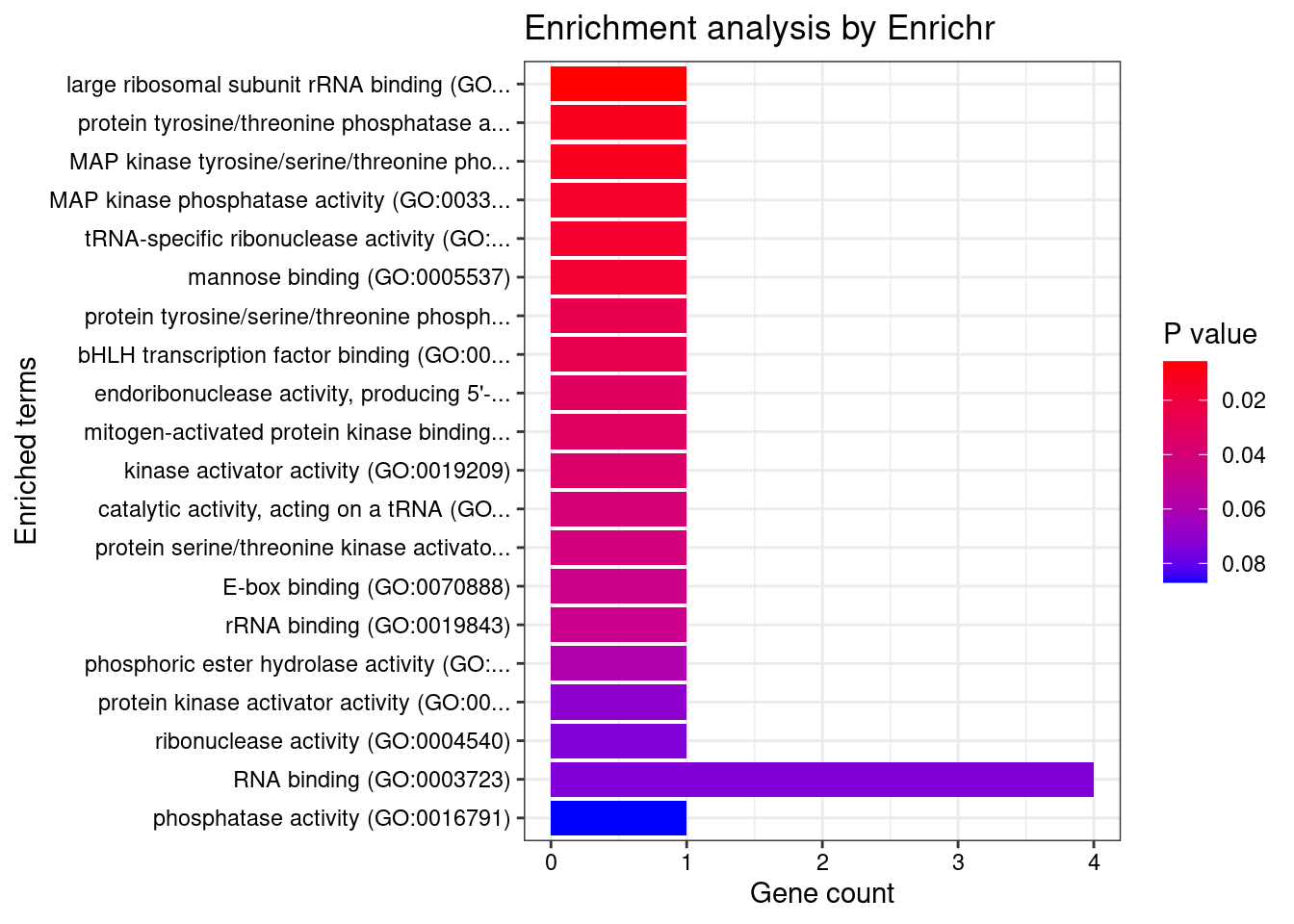
[1] Term Overlap Adjusted.P.value Genes
<0 rows> (or 0-length row.names)DisGeNET enrichment analysis for genes with PIP>0.5
Description FDR Ratio BgRatio
29 Prostatic Neoplasms 0.02411 4/9 616/9703
45 Malignant neoplasm of prostate 0.02411 4/9 616/9703
62 Refractory anemia with ringed sideroblasts 0.02411 1/9 2/9703
74 PROSTATE CANCER, HEREDITARY, 2 0.02411 1/9 1/9703
76 COMBINED OXIDATIVE PHOSPHORYLATION DEFICIENCY 17 0.02411 1/9 1/9703
78 MENTAL RETARDATION, AUTOSOMAL RECESSIVE 52 0.02411 1/9 1/9703
41 Malignant mesothelioma 0.03368 2/9 109/9703
42 Malignant melanoma of iris 0.03368 1/9 5/9703
43 Malignant melanoma of choroid 0.03368 1/9 5/9703
55 Long Sleeper Syndrome 0.03368 1/9 7/9703WebGestalt enrichment analysis for genes with PIP>0.5
Loading the functional categories...
Loading the ID list...
Loading the reference list...
Performing the enrichment analysis...Warning in oraEnrichment(interestGeneList, referenceGeneList, geneSet, minNum =
minNum, : No significant gene set is identified based on FDR 0.05!NULLPIP Manhattan Plot
Sensitivity, specificity and precision for silver standard genes
#number of genes in known annotations
print(length(known_annotations))[1] 130#number of genes in known annotations with imputed expression
print(sum(known_annotations %in% ctwas_gene_res$genename))[1] 49#significance threshold for TWAS
print(sig_thresh)[1] 4.553#number of ctwas genes
length(ctwas_genes)[1] 8#number of TWAS genes
length(twas_genes)[1] 113#show novel genes (ctwas genes with not in TWAS genes)
ctwas_gene_res[ctwas_gene_res$genename %in% novel_genes,report_cols] genename region_tag susie_pip mu2 PVE z num_eqtl
10112 TMEM222 1_19 0.8254 22.21 0.0001740 4.303 1
3590 BHLHE41 12_18 0.8795 24.00 0.0002004 4.516 1#sensitivity / recall
print(sensitivity) ctwas TWAS
0.01538 0.11538 #specificity
print(specificity) ctwas TWAS
0.9994 0.9896 #precision / PPV
print(precision) ctwas TWAS
0.2500 0.1327 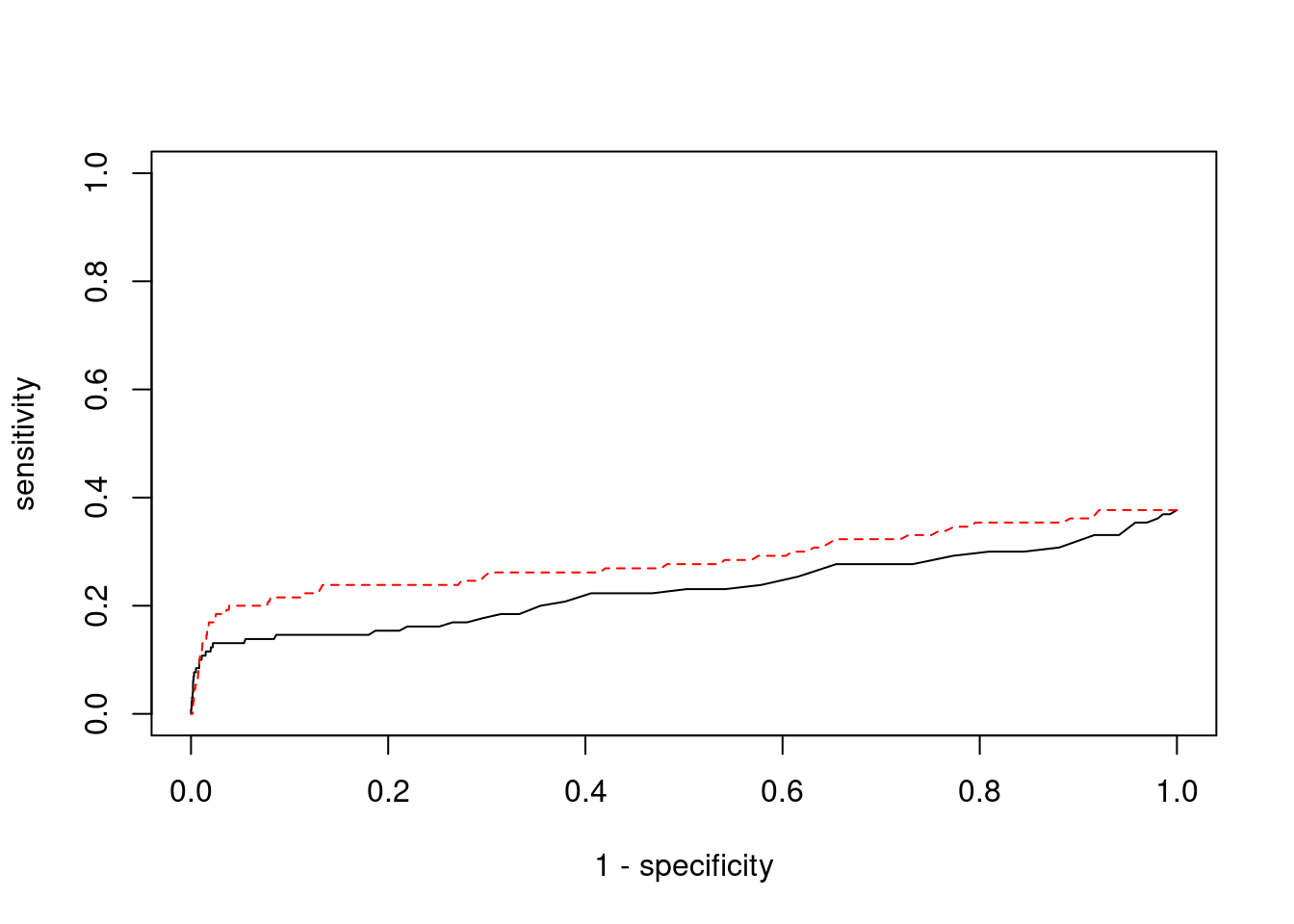
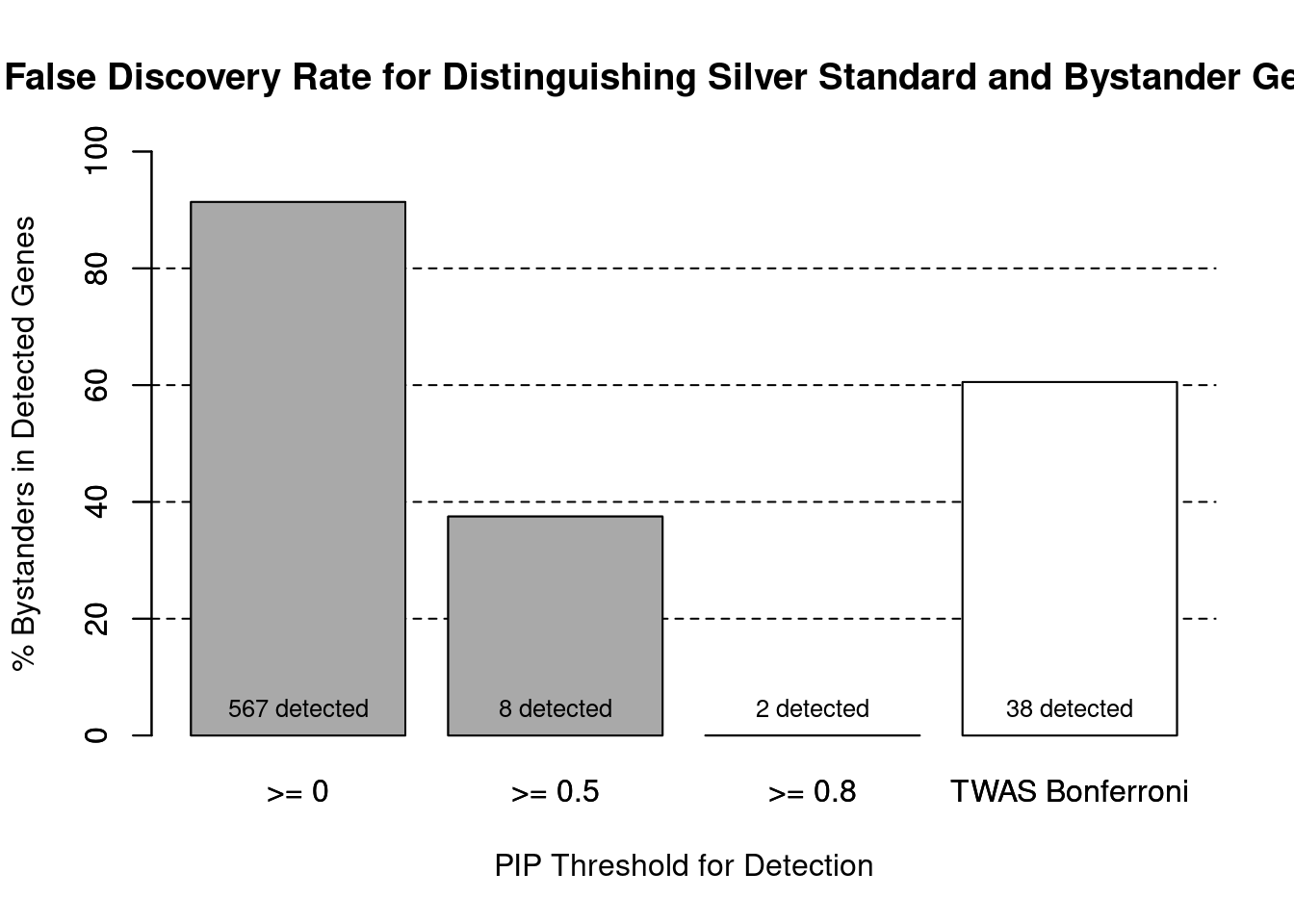
cTWAS is more precise than TWAS in distinguishing silver standard and bystander genes
#number of genes in known annotations (with imputed expression)
print(length(known_annotations))[1] 49#number of bystander genes (with imputed expression)
print(length(unrelated_genes))[1] 518#subset results to genes in known annotations or bystanders
ctwas_gene_res_subset <- ctwas_gene_res[ctwas_gene_res$genename %in% c(known_annotations, unrelated_genes),]
#assign ctwas and TWAS genes
ctwas_genes <- ctwas_gene_res_subset$genename[ctwas_gene_res_subset$susie_pip>0.8]
twas_genes <- ctwas_gene_res_subset$genename[abs(ctwas_gene_res_subset$z)>sig_thresh]
#significance threshold for TWAS
print(sig_thresh)[1] 4.553#number of ctwas genes (in known annotations or bystanders)
length(ctwas_genes)[1] 2#number of TWAS genes (in known annotations or bystanders)
length(twas_genes)[1] 38#sensitivity / recall
sensitivity ctwas TWAS
0.04082 0.30612 #specificity / (1 - False Positive Rate)
specificity ctwas TWAS
1.0000 0.9556 #precision / PPV / (1 - False Discovery Rate)
precision ctwas TWAS
1.0000 0.3947 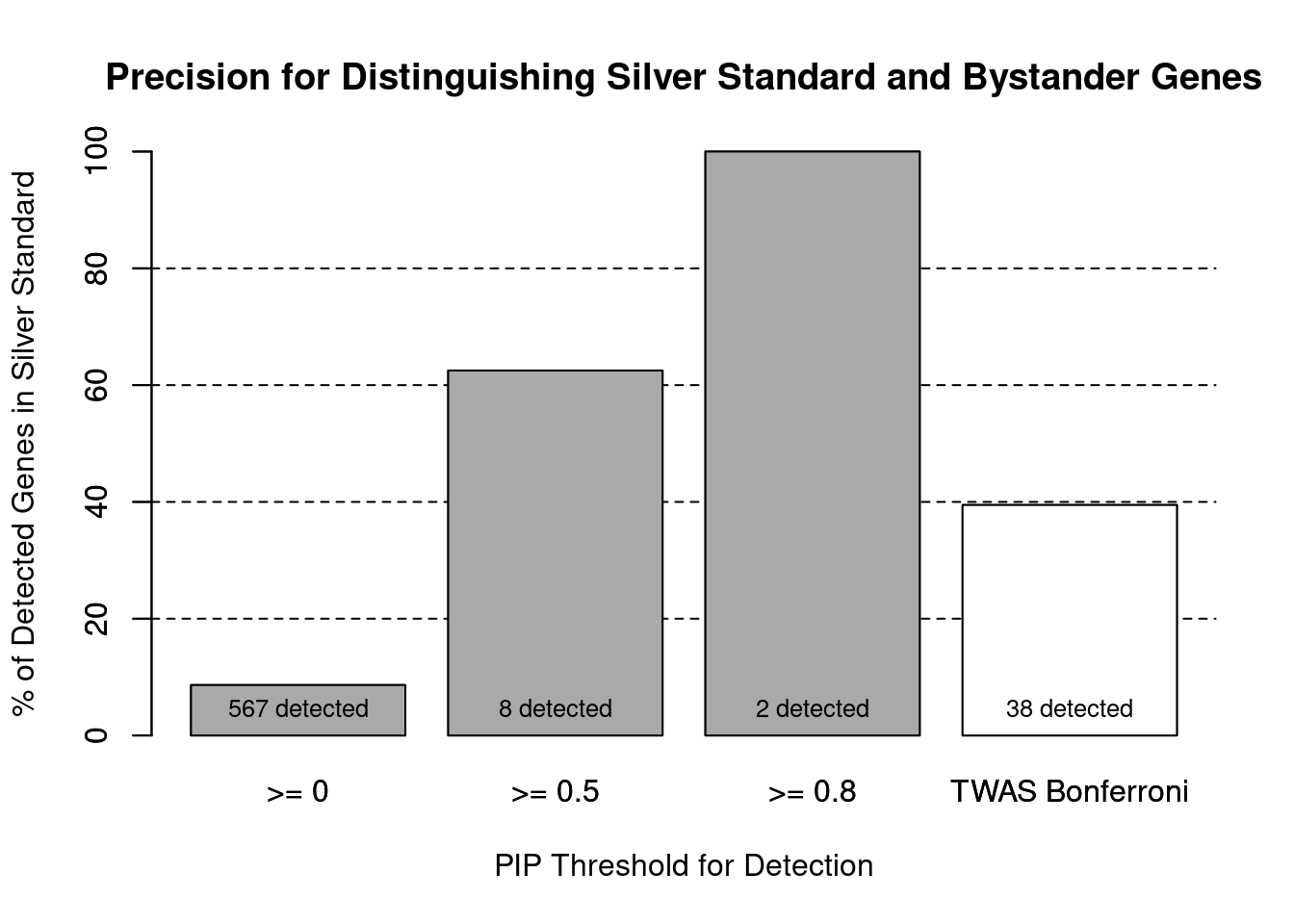
pip_range <- (0:1000)/1000
sensitivity <- rep(NA, length(pip_range))
specificity <- rep(NA, length(pip_range))
for (index in 1:length(pip_range)){
pip <- pip_range[index]
ctwas_genes <- ctwas_gene_res_subset$genename[ctwas_gene_res_subset$susie_pip>=pip]
sensitivity[index] <- sum(ctwas_genes %in% known_annotations)/length(known_annotations)
specificity[index] <- sum(!(unrelated_genes %in% ctwas_genes))/length(unrelated_genes)
}
plot(1-specificity, sensitivity, type="l", xlim=c(0,1), ylim=c(0,1), main="", xlab="1 - Specificity", ylab="Sensitivity")
title(expression("ROC Curve for cTWAS (black) and TWAS (" * phantom("red") * ")"))
title(expression(phantom("ROC Curve for cTWAS (black) and TWAS (") * "red" * phantom(")")), col.main="red")
sig_thresh_range <- seq(from=0, to=max(abs(ctwas_gene_res_subset$z)), length.out=length(pip_range))
for (index in 1:length(sig_thresh_range)){
sig_thresh_plot <- sig_thresh_range[index]
twas_genes <- ctwas_gene_res_subset$genename[abs(ctwas_gene_res_subset$z)>=sig_thresh_plot]
sensitivity[index] <- sum(twas_genes %in% known_annotations)/length(known_annotations)
specificity[index] <- sum(!(unrelated_genes %in% twas_genes))/length(unrelated_genes)
}
lines(1-specificity, sensitivity, xlim=c(0,1), ylim=c(0,1), col="red", lty=1)
abline(a=0,b=1,lty=3)
#add previously computed points from the analysis
ctwas_genes <- ctwas_gene_res_subset$genename[ctwas_gene_res_subset$susie_pip>0.8]
twas_genes <- ctwas_gene_res_subset$genename[abs(ctwas_gene_res_subset$z)>sig_thresh]
points(1-specificity_plot["ctwas"], sensitivity_plot["ctwas"], pch=21, bg="black")
points(1-specificity_plot["TWAS"], sensitivity_plot["TWAS"], pch=21, bg="red")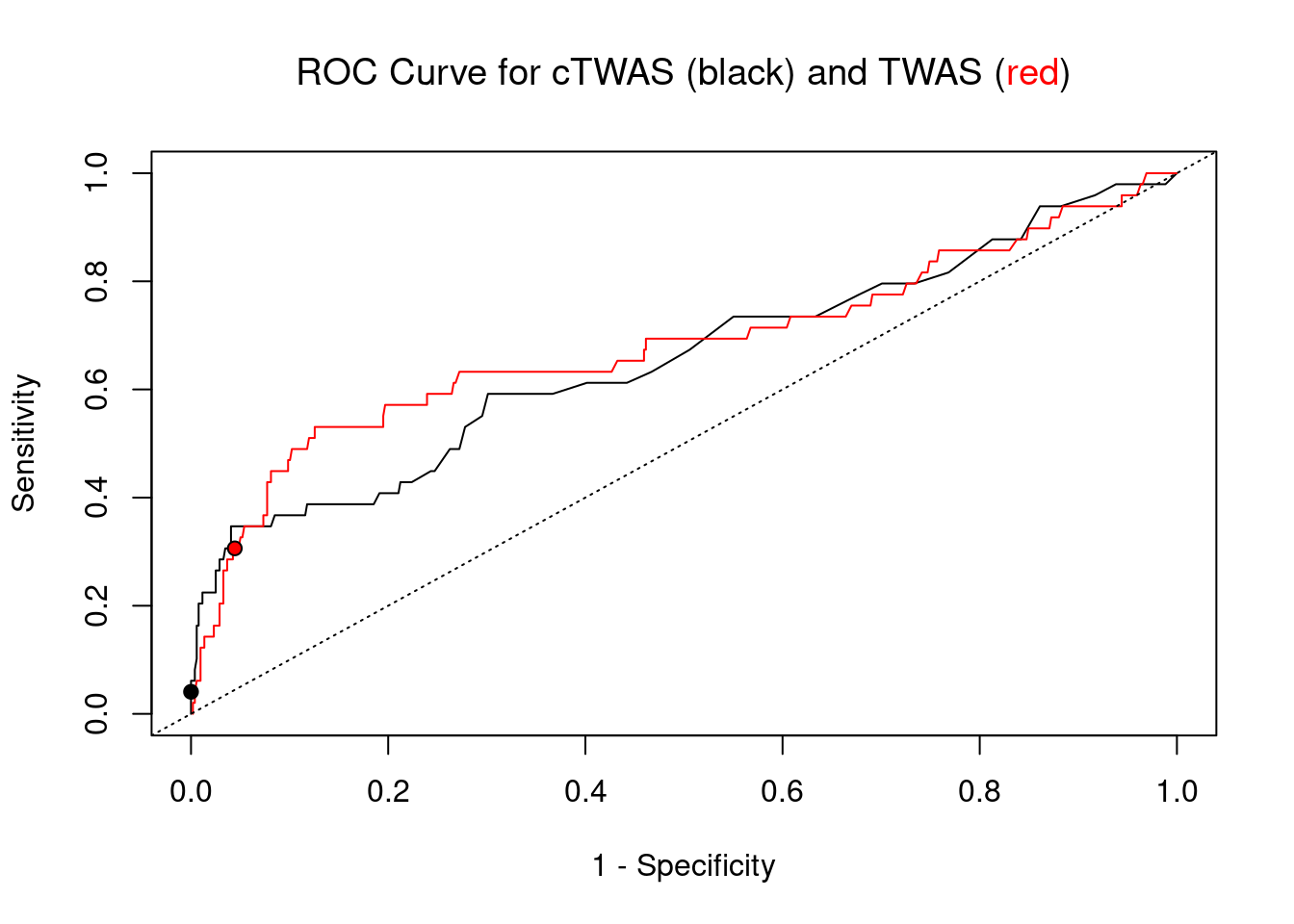
Undetected silver standard genes have low TWAS z-scores or stronger signal from nearby variants
#table of outcomes for silver standard genes
-sort(-table(silver_standard_case))silver_standard_case
Not Imputed Insignificant z-score Nearby SNP(s)
81 34 13
Detected (PIP > 0.8)
2 #show inconclusive genes
silver_standard_case[silver_standard_case=="Inconclusive"]named character(0)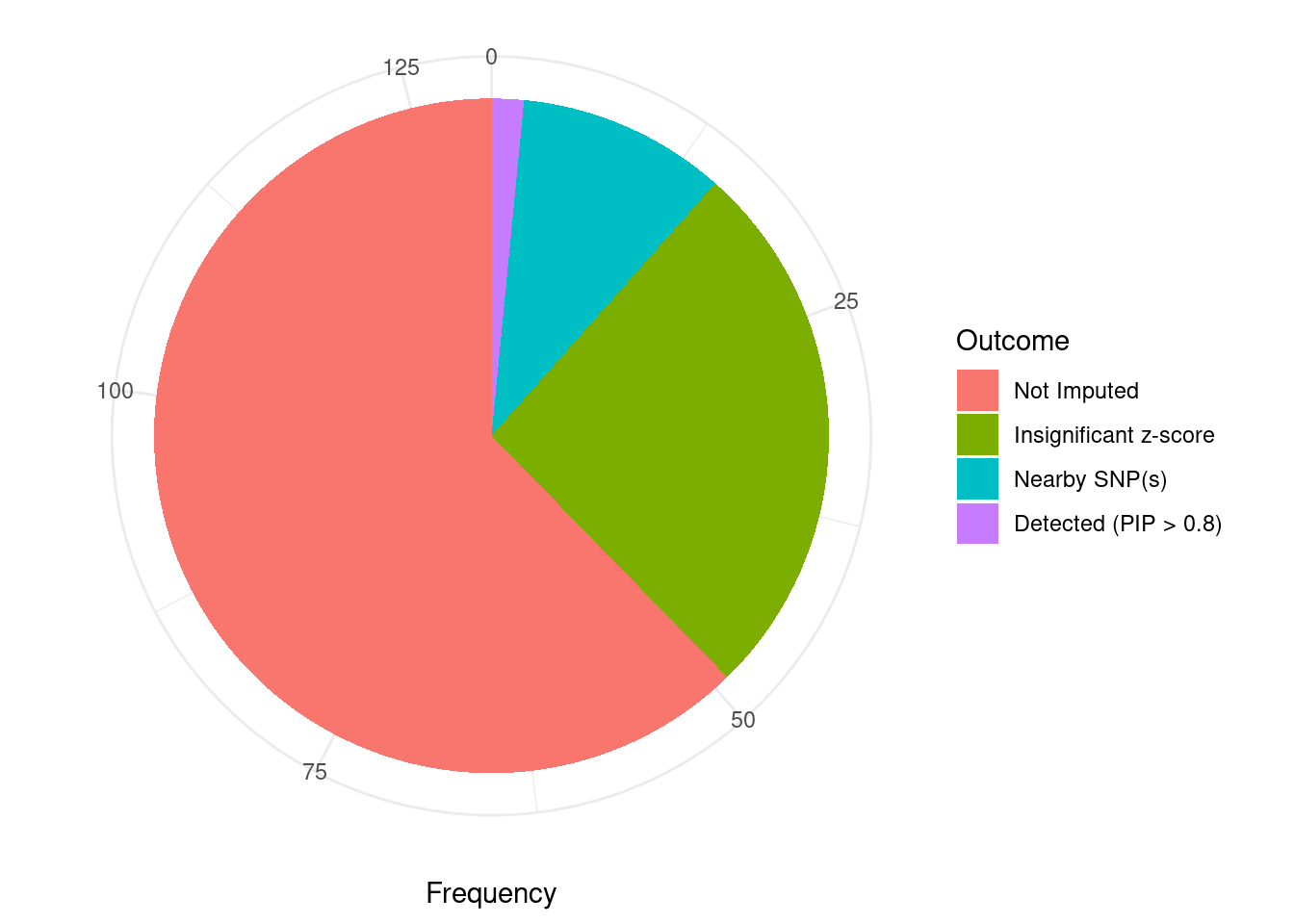
sessionInfo()R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] GenomicRanges_1.36.1 GenomeInfoDb_1.20.0 IRanges_2.18.1
[4] S4Vectors_0.22.1 BiocGenerics_0.30.0 biomaRt_2.40.1
[7] readxl_1.3.1 forcats_0.5.1 stringr_1.4.0
[10] dplyr_1.0.7 purrr_0.3.4 readr_2.1.1
[13] tidyr_1.1.4 tidyverse_1.3.1 tibble_3.1.6
[16] WebGestaltR_0.4.4 disgenet2r_0.99.2 enrichR_3.0
[19] cowplot_1.1.1 ggplot2_3.3.5 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] ggbeeswarm_0.6.0 colorspace_2.0-2 rjson_0.2.20
[4] ellipsis_0.3.2 rprojroot_2.0.2 XVector_0.24.0
[7] fs_1.5.2 rstudioapi_0.13 farver_2.1.0
[10] ggrepel_0.9.1 bit64_4.0.5 AnnotationDbi_1.46.0
[13] fansi_1.0.2 lubridate_1.8.0 xml2_1.3.3
[16] codetools_0.2-16 doParallel_1.0.17 cachem_1.0.6
[19] knitr_1.36 jsonlite_1.7.2 apcluster_1.4.8
[22] Cairo_1.5-12.2 broom_0.7.10 dbplyr_2.1.1
[25] compiler_3.6.1 httr_1.4.2 backports_1.4.1
[28] assertthat_0.2.1 Matrix_1.2-18 fastmap_1.1.0
[31] cli_3.1.0 later_0.8.0 prettyunits_1.1.1
[34] htmltools_0.5.2 tools_3.6.1 igraph_1.2.10
[37] GenomeInfoDbData_1.2.1 gtable_0.3.0 glue_1.6.2
[40] reshape2_1.4.4 doRNG_1.8.2 Rcpp_1.0.8
[43] Biobase_2.44.0 cellranger_1.1.0 jquerylib_0.1.4
[46] vctrs_0.3.8 svglite_1.2.2 iterators_1.0.14
[49] xfun_0.29 ps_1.6.0 rvest_1.0.2
[52] lifecycle_1.0.1 rngtools_1.5.2 XML_3.99-0.3
[55] zlibbioc_1.30.0 getPass_0.2-2 scales_1.1.1
[58] vroom_1.5.7 hms_1.1.1 promises_1.0.1
[61] yaml_2.2.1 curl_4.3.2 memoise_2.0.1
[64] ggrastr_1.0.1 gdtools_0.1.9 stringi_1.7.6
[67] RSQLite_2.2.8 highr_0.9 foreach_1.5.2
[70] rlang_1.0.1 pkgconfig_2.0.3 bitops_1.0-7
[73] evaluate_0.14 lattice_0.20-38 labeling_0.4.2
[76] bit_4.0.4 processx_3.5.2 tidyselect_1.1.1
[79] plyr_1.8.6 magrittr_2.0.2 R6_2.5.1
[82] generics_0.1.1 DBI_1.1.2 pillar_1.6.4
[85] haven_2.4.3 whisker_0.3-2 withr_2.4.3
[88] RCurl_1.98-1.5 modelr_0.1.8 crayon_1.5.0
[91] utf8_1.2.2 tzdb_0.2.0 rmarkdown_2.11
[94] progress_1.2.2 grid_3.6.1 data.table_1.14.2
[97] blob_1.2.2 callr_3.7.0 git2r_0.26.1
[100] reprex_2.0.1 digest_0.6.29 httpuv_1.5.1
[103] munsell_0.5.0 beeswarm_0.2.3 vipor_0.4.5